

Reactive Intermediate Chemistry
.pdf

346 STABLE SINGLET CARBENES |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
R2P |
|
C |
|
|
|
N |
i-Pr |
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
i-Pr |
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
R2P |
|
|
|
|
i-Pr |
LiHMDS |
|
R2P |
|
|
|
|
|
|
|
|
|
|
i-Pr |
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||
C |
|
N |
X − |
|
|
|
|
|
|
|
C |
|
|
|
N |
|
XVa-d |
|||||||||
|
|
|
|
|
|
|
|
|||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||
|
−78 °C |
|
|
|
|
|
|
|
|
|
||||||||||||||||
H |
i-Pr |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
i-Pr |
||||||||
15a-d |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
a: R = c-Hex2N, b: R = i-Pr2N, |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
|
R2P |
|
|
|
|
|
|
|
|
|
|
i-Pr |
|
|||||||||||||
c: R = Ph, d: R = t-Bu |
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
|
C |
|
|
|
|
N |
||||||||||||||||||||
X = CF3SO3 |
|
|
|
|
|
|
|
|
|
|
i-Pr |
|
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Scheme 8.10
methylcarbene (XIVc) could be observed by 31P NMR up to 50 C (t1=2 10 min at 50 C), where it quickly isomerizes (Scheme 8.9).
As we have seen, all known stable singlet carbenes feature either an amino or a phosphino substituent. The preparation of carbenes XIV demonstrates that the presence of a phosphino substituent allows for the spectroscopic characterization of singlet carbenes under standard laboratory conditions, when the steric bulk of the spectator substituent is decreased even to the size of a methyl group. Therefore, it was of interest to find out if a phosphino group was more efficient for stabilizing a carbene center than an amino group, or vice versa. The latter situation would imply that a broad range of stable monoaminocarbenes would be easily available. We approached this problem by synthesizing the (phosphino)(amino)carbenes (XV).67
Carbenes XVa–d were cleanly generated at 78 C by deprotonation of the corresponding phosphinoiminium salts 15a–d with the lithium salt of hexamethyldisilylazane (Scheme 8.10), and were characterized by multinuclear NMR spectroscopy at 30 C. The main feature of the NMR spectra of XVa–d is the very low field values of the 13C chemical shifts of the carbene carbon atoms (d: 320–348 ppm, JPC: 22–101 Hz). These signals are even at lower field than those observed for the other known aminocarbenes (210–300 ppm) and in a totally different range from those for phosphinocarbenes (70–180 ppm). In all cases, 1H NMR spectra showed the presence of two different isopropyl groups on the nitrogen atom bound to the carbene center, which indicates the absence of rotation about the C N bond. All these NMR data strongly suggest that only the amino substituent interacts with the carbene center, the phosphino group merely remaining a spectator substituent.
In the case of XVa (R ¼ c-Hex2N), orange crystals suitable for an X-ray diffrac-
|
C (Fig. 8.12). |
|
tion study were obtained by cooling a saturated ether solution to 30 |
||
˚ |
¼ |
304.5 ) and |
The pyramidalization of the phosphorus atom (sum of bond angles |
|
|
the long P1C1 bond length (1.856 A), which is in the range for PC single bonds, demonstrate that the phosphino group is indeed a spectator substituent. As

|
|
REACTIVITY OF STABLE SINGLET CARBENES |
347 |
||||
|
|
(c-Hex)2N |
P |
|
P |
i-Pr |
|
P1 |
N1 |
C N |
|
(c-Hex)2N |
C N |
|
|
N2A |
(c-Hex)2N |
|
i-Pr |
(c-Hex)2N |
i-Pr |
|
|
|
|
|
|||||
N2 |
|
|
|
||||
|
|
|
i-Pr |
|
|
|
|
C1
XVa
Figure 8.12. Molecular structure of carbene XVa.
expected, the nitrogen atom is in a planar environment (sum of bond angles ¼
˚
359.6 ), and the N1 C1 bond length (1.296 A) is short. Finally, the carbene bond angle is small (116.5 ) as expected for aminocarbenes and in contrast with (phosphino)carbenes.
The stability of carbenes XVa–d is dependent on the nature of the phosphorus substituents. All the carbenes are stable for days at T < 20 C; the most stable is XVd (R ¼ t-Bu), which can be stored in solution for a few days at 20 C.
From these data, it can be stated that an amino group is much more efficient for stabilizing a carbene center than a phosphino group. These results also resolve a controversy: If an aminocarbene is considered a carbene despite the interaction between the amino group and the carbene center, a phosphinocarbene should also be considered a carbene.
5. REACTIVITY OF STABLE SINGLET CARBENES
Whereas triplet carbenes exhibit radical-like reactivity, singlet carbenes are expected to show nucleophilic as well as electrophilic behavior because of the lone pair and vacant orbital. This section will be focused on the most typical reactions involving stable singlet carbenes: dimerizations, cyclopropanations, and formation of ylides and reverse ylides with Lewis bases and acids, respectively. Since, as recently noted by Herrmann:68 ‘‘a revolutionary turning point in organometallic catalysis is emerging,’’ a part of this section will devoted to the role of stable singlet carbenes as ligands for transition metal catalysts.
5.1. Dimerization
First, it should be repeated that despite early69a and recent claims,69b no examples of thermal dissociation of alkenes into carbenes (the reverse reaction of the carbene dimerization) are known.70 However, in their recent paper, Lemal and co-worker70 pointed out that electrophiles can catalyze the dissociation of tetraaminoalkenes.
In the Carter and Goddard formulation,71 the strength of the C C double bond resulting from the dimerization of singlet carbenes should correspond to that of a canonical C C double bond (usually that of ethene) minus twice the singlet–triplet

348 STABLE SINGLET CARBENES
N N N N
Na
 N N
N N
 N N
N N
{IVd}2
N N N N
Na
 N N
N N
 N N
N N
IVe
Scheme 8.11
energy gap for the carbene. For example, the singlet–triplet splitting in the parent imidazol-2-ylidene (IV) has been calculated to be 85 kcal/mol;72 accordingly, one expects the CC bond strength in the dimer to be approximately only ½172 ð2 85Þ& ¼ 2 kcal/mol. A convincing experimental proof of the weakness of the C C bond in tetraamino alkenes comes from the very elegant work by Taton and Chen.73 The tightly constrained {IVd}2 exists as the tetraazafulvalene, whereas the homologue with longer tethers dissociates into a biscarbene (IVe, Scheme 8.11). Perhaps equally remarkable is the fact that the central C C bond in {IVd}2 is of
˚
normal length (1.337 A), even though the strength of this bond can be only a few kilocalories per mole.
Similarly to imidazol-2-ylidene (IV), the calculated value for the energy of dimerization of Enders-type carbene VII is only 9.5 kcal/mol.74 These remarkably small values, at least partially due to the loss of aromaticity in the carbene dimers {IV}2 and {VII}2, highlight the difficulty of dimerization of such carbenes. In contrast, in the case of the parent acyclic diaminocarbene IX, Heinemann and Thiel72 found a dimerization energy of 45 kcal/mol. This poses another question What is the value of the energy barrier for the dimerization? Nowadays, the dimerization of singlet carbenes is believed to follow a nonleast motion pathway that involves
the attack of the occupied in-plane s lone pair of one singlet carbene center on the out-of-plane vacant pp orbital of a second carbene (Fig. 8.13).71,75 Calculations
pπ σ
 C C
C C
σ pπ
Figure 8.13. Schematic representation of the mechanism for the dimerization of singlet carbenes.
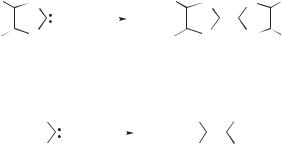
|
|
|
|
REACTIVITY OF STABLE SINGLET CARBENES 349 |
|||||||||||||||||||
|
Dipp |
|
|
|
|
|
|
|
|
|
|
|
|
Dipp |
|
|
|
|
|
||||
Me |
|
|
|
|
|
|
|
|
|
|
|
|
Me |
|
|
|
|
|
|
S |
Me |
||
N |
|
H |
|
|
|
|
|
|
|
|
|
N |
|
||||||||||
2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
S |
cat. |
|
|
S |
|
|
|
|
|
||||||||||||||
Me |
Me |
|
|
N |
Me |
||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
VIIIa |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Dipp |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
{VIIIa}2 |
|
|||||||||
Dipp = 2,6-(i-Pr)2C6H3 |
|
|
|
|
|
|
|
|
|
|
|
||||||||||||
2 |
R2N |
|
H |
|
|
|
|
|
R2N |
NR2 |
|
||||||||||||
|
|
|
|
|
|
|
|||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
R2N |
|
cat. |
|
R2N |
NR2 |
|
|||||||||||||||||
|
|
|
|
||||||||||||||||||||
|
IXb,c |
|
|
|
|
|
|
|
|
|
|
|
|
{IXb,c}2 |
|
||||||||
b: R2 = piperidinyl; c: R = Me
Scheme 8.12
regarding the dimerization path indicate a significant barrier of 19.4 kcal/mol for the parent 1,2,4-triazol-5-ylidene (VII),74 and Alder estimated the G for the
dimerization of the bis(N-piperidinyl)carbene (IXb) and bis(dimethylamino)carbene (IXc) to be >25 kcal/mol.53,76 These large values are not surprising because
the dimerization reaction involves the carbene vacant orbital that is very high in energy due to the donation of the nitrogen lone pairs.
However, besides the work by Wanzlick, several examples of aminocarbene dimerizations have been reported.43,53,77 Noteworthy is the isolation of both the thiazol-2-ylidene (VIIIa) and of its dimer {VIIIa}243 and the spectroscopic characterization by Alder and Blake of the bis(N-piperidinyl)carbene (IXb) and of the tetrakis(N-piperidinyl)ethene ({IXb}2)53 (Scheme 8.12).
Interestingly, Arduengo observed that VIIIa was stable with respect to dimerization in the absence of a Brønsted or Lewis acid catalyst.43 Similarly, in the absence of an acid catalyst, dimerization of IXb is extremely slow and is first order in carbene.53 Therefore, the observed formal dimerization of VIIIa and IXb,c does not involve the coupling of two carbenes, but the nucleophilic attack of one carbene upon its conjugate acid, followed by proton elimination, as already suggested by Chen and Jordan78 (Scheme 8.13). It is important to keep in mind that even N,N-dialkylimidazolium ions have pKa values of 24 in DMSO,79 and based on the calculated proton acidity, Alder estimated the pKa values for acyclic diaminocarbenes to be from 2 to 6 pKa units higher than for imidazol-2-ylidenes.54
Another important issue has recently been raised by Alder. He proved that diaminocarbenes, including aromatic carbenes, coordinate alkali metal ions (coming from the base), which is important with regard to the rate of dimerization.40 The metal ions might act as a Lewis acid catalyst for dimerization, as observed for protons, but alternatively, strong complexation might stabilize the carbene center and prevent its dimerization. This question is still open to debate.

350 STABLE SINGLET CARBENES
|
|
|
|
|
|
|
|
|
|
|
|
R2N |
|
|
|
|
|
|
|
|
R2N |
|||||||||
R2N |
H |
|
|
|
|
R |
N |
|
R |
N |
R |
N |
NR |
2 |
|
R |
N |
|||||||||||||
|
|
|
||||||||||||||||||||||||||||
|
2 |
|
|
|
2 |
|
|
|
|
|
|
2 |
|
|
|
H |
2 |
|
|
|||||||||||
R2N |
|
|
|
|
|
|
|
|
|
|
H |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
R2N |
|
|
|
|
|
|
|
R2N |
|
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
NR2 |
|
|
|
|
|
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
R2N |
|
|
|
|
|
R2N |
|
|
|
NR2 |
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
H |
+ |
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
R2N |
|
|
|
|
|
R2N |
|
|
|
NR2 |
|||||||
Scheme 8.13
Only one example of dimerization of a phosphinocarbene has been reported. As already briefly mentioned, carbene XIIa is stable for days in solution at 30 C in THF or toluene solution, but in pentane or upon evaporation of the donor solvent, even at 50 C, a dimerization generating exclusively the alkene {XIIa}2 is observed.56 Interestingly, upon warming the THF solution of XIIa to 20 C, a clean rearrangement occurs affording a cumulene, with no trace of the carbene dimer {XIIa}2 (Scheme 8.14). These observations are in perfect agreement with previous work on transient carbenes. It has been shown that the extent of 1,2-migra- tion processes increases, relative to intermolecular reaction (like the dimerization), as the solvent is changed from an alkane to a donor solvent.80
|
|
|
|
pentane |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
or |
F3C |
|
|
|
|
PR2 |
|
|
|
|
|
|
|
|
|
|
|
hex-1-ene |
C |
|
C |
|
|
|
|
|
|
|
|||
|
|
|
|
|
R2P |
|
CF3 |
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
R2P CF3 |
hν |
|
|
{XIIa}2 |
|
|
|
|
|
|
|
||||||
C |
−60 °C |
THF |
−50 |
°C |
|
evaporation |
|||||||||||
|
|
|
ether |
|
|||||||||||||
N2 |
|
|
of the solvent |
||||||||||||||
|
|
|
|
|
|
||||||||||||
R = c-Hex2N |
|
or |
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
R2P CF3 |
−20 °C |
|||||||||||||||
|
toluene |
||||||||||||||||
|
|
|
|
||||||||||||||
|
|
|
|
|
|
|
XIIa |
|
R2P |
|
C |
|
CF2 |
||||
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
F |
|||||||||
Scheme 8.14
5.2. Cyclopropanation
In a recent paper,58c Moss wrote: ‘‘The addition of a carbene to an alkene with the formation of a cyclopropane is perhaps the most fundamental of cycloaddition reactions, as well as a basic component of the synthetic armamentarium.’’ Indeed, cyclopropanation reactions involving transient carbenes81 or even transition metal carbene complexes have been widely studied.82 Both singlet and triplet transient carbenes undergo cyclopropanation reactions, although with a totally different

|
|
|
|
|
|
|
|
|
REACTIVITY OF STABLE SINGLET CARBENES |
351 |
||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
Ph |
|
|
|
|
N |
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
Ph |
|
|
|
N |
|
|
|
|
N |
|
|
Ph |
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
MeO2C |
|
|
|
|
CO2Me |
|
|
|
|||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
|
|
|
|
|
|
|
|
|
CO2Me |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Ph |
MeO2C |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
N |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
Ph |
|
N N |
|
Ph |
|
or |
|
|
Ph |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||
|
|
|
|
|
|
|
MeO2C |
|
CO2Me |
|
|
|
|
|
|
|
|
N |
|
|
|
Ph |
|
|
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
N |
|||||||||||||||
|
|
VIIa |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
Ph |
|
|
N |
|
|
|
|
N |
|
|
Ph |
N |
|
Ph |
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
H |
|
|
Ph |
|
N |
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CO2Me |
|
|
CO2Me |
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CO2Me |
|
|
|
|
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CO2Me |
|||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
54 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
Scheme 8.15
mechanism, which is apparent from the stereochemistry of the reaction: with singlet carbenes the stereochemistry about the original carbon–carbon double bond is maintained, while with triplet carbenes the stereochemical information is lost.83
1,2,4-Triazol-5-ylidene (VIIa) reacts with diethyl fumarate and also diethyl maleate, not giving the corresponding cyclopropane, but the methylenetriazoline derivative (Scheme 8.15).42 According to Enders et al.,42 a ½1 þ 2&-cycloaddition first occurs, leading to the transient cyclopropane. Then, a ring opening would lead to a zwitterionic derivative, which would undergo a 1,2-H shift [AM1 calculations predict a strongly negative reaction enthalpy ( H ¼ 18 kcal/mol) for the rearrangement of the cyclopropane to the isolated product].42 However, it is quite clear that a mechanism directly leading to the zwitterionic species also explains the experimental results.
In marked contrast, we used the cyclopropanation reaction of (phosphino)(silyl)- carbene (Ia) with methyl acrylate to demonstrate the carbene nature of our compound.5b More recently, we studied in detail the stereochemistry of this type of reaction.84 The (phosphino)(silyl)carbene (Ia) reacted efficiently with 3,3,4,4,5, 5,6,6,6-nonafluorohex-1-ene and (Z)- and (E)-2-deuteriostyrene giving the corresponding cyclopropanes in good yields. The stereochemical outcome was such that all monosubstituted alkenes gave exclusively the syn isomer (with respect to the phosphino group), and the addition of disubstituted alkenes was totally stereospecific (Scheme 8.16).
The stereospecificity observed with (Z)- and (E)-2-deuteriostyrene presents convincing evidence for the concerted nature of the cyclopropanation reaction, and therefore the genuine singlet carbene nature of stable (phosphino)(silyl)carbenes (I). The diastereoselectivity is at first glance surprising. Indeed, it is clear that steric factors cannot govern the observed selectivity since the bis(amino)phosphino group


 C
C 


