Reactive Intermediate Chemistry
.pdf324SINGLET CARBENES
55.N. J. Turro, J. A. Butcher, Jr., R. A. Moss, W. Guo, R. C. Munjal, and M. Fedorynski,
J.Am. Chem. Soc. 1980, 102, 7576.
56.J. J. Zupanic and G. B. Schuster, J. Am. Chem. Soc. 1980, 102, 5958.
57.See R. A. Moss and N. J. Turro, in Ref. 9, pp. 213ff.
58.D. P. Cox, I. R. Gould, N. P. Hacker, R. A. Moss, and N. J. Turro, Tetrahedron Lett. 1983, 24, 5313.
59.R. A. Moss, H. Fan, L. M. Hadel, S. Shen, J. Wlostowska, M. Wlostowski, and K. KroghJespersen, Tetrahedron Lett. 1987, 28, 4779.
60.R. A. Moss, W. Lawrynowicz, L. M. Hadel, N. P. Hacker, N. J. Turro, I. R. Gould, and
Y.Cha, Tetrahedron Lett. 1986, 21, 4125.
61.R. A. Moss, C.-S. Ge, J. Wlostowska, E. G. Jang, E. A. Jefferson, and H. Fan,
Tetrahedron Lett. 1995, 36, 3083.
62.R. A. Moss, S. Shen, L. M. Hadel, G. Kmiecik-Lawrynowicz, J. Wlostowska, and
K.Krogh-Jespersen, J. Am. Chem. Soc. 1987, 109, 4341.
63.For further discussions of C6H5CX reactivity, see I. R. Gould, N. J. Turro, J. Butcher, Jr.,
C.Doubleday, Jr., N. P. Hacker, G. F. Lehr, R. A. Moss, D. P. Cox, W. Guo, R. C. Munjal,
L.A. Perez, and M. Fedorynski, Tetrahedron 1985, 41, 1587.
64.J. E. Chateauneuf, R. P. Johnson, and M. M. Kirchhoff, J. Am. Chem. Soc. 1990, 112, 3217.
65.J. E. Jackson, N. Soundararajan, M. S. Platz, and M. T. H. Liu, J. Am. Chem. Soc. 1988, 110, 5595. See J. E. Jackson and M. S. Platz, in Ref. 10, pp. 89ff; M. S. Platz, in Ref. 13, pp. 27ff.
66.N. J. Turro, G. F. Lehr, J. A. Butcher, Jr., R. A. Moss, and W. Guo, J. Am. Chem. Soc. 1982, 104, 1754.
67.(a) K. N. Houk, N. G. Rondan, and J. Mareda, J. Am. Chem. Soc. 1984, 106, 4293;
(b) Tetrahedron 1985, 41, 1555.
68.B. Giese and C. Neumann, Tetrahedron Lett. 1982, 23, 3357. B. Giese, W.-B. Lee, and
C.Neumann, Angew. Chem. Int. Ed. 1982, 21, 310.
69.R. A. Moss, W. Lawrynowicz, N. J. Turro, I. R. Gould, and Y. Cha, J. Am. Chem. Soc. 1986, 108, 7028.
70.N. J. Turro, M. Okamoto, I. R. Gould, R. A. Moss, W. Lawrynowicz, and L. M. Hadel,
J.Am. Chem. Soc. 1987, 109, 4973.
71.W. R. Moore, W. R. Moser, and J. E. LaPrade, J. Org. Chem. 1963, 28, 2200.
72.B. Zurawski and W. Kutzelnigg, J. Am. Chem. Soc. 1978, 100, 2654; J. F. Blake,
S.G. Wiershke, and W. L. Jorgensen, J. Am. Chem. Soc. 1989, 111, 1919; A. E. Keating,
M.A. Garcia-Garibay, and K. N. Houk, J. Am. Chem. Soc. 1997, 119, 10805.
73.A. E. Keating, S. R. Merrigan, D. A. Singleton, and K. N. Houk, J. Am. Chem. Soc. 1999, 121, 3933.
74.Structures 20 and 21 reproduced from Ref. 73 with permission from A. E. Keating,
S.R. Merrigan, and K. N. Houk, J. Am. Chem. Soc., 1999, 121, 3933. Copyright #1999 the American Chemical Society.
75.See Ref. 35 (Hoffmann, Hayes, and Skell), in which such geometries are also analyzed.
76.G. L. Closs and L. E. Closs, Angew. Chem. 1962, 74, 431; G. L. Closs and R. A. Moss,
J.Am. Chem. Soc. 1964, 86, 4042.
77.G. Boche and J. C. W. Lohrenz, Chem. Rev. 2001, 101, 697.
REFERENCES 325
78.W. von E. Doering and P. LaFlamme, J. Am. Chem. Soc. 1956, 78, 5447.
79.M. Jones, Jr., W. Ando, M. E. Hendrick, A. Kulczycki, Jr., P. M. Howley, K. R. Hummel, and D. S. Malament, J. Am. Chem. Soc. 1972, 94, 7469.
80.N. J. Turro, Y. Cha, and I. R. Gould, J. Am. Chem. Soc. 1987, 109, 2101.
81.K. R. Kopecky, G. S. Hammond, and P. A. Leermakers, J. Am. Chem. Soc. 1962, 84, 1015.
82.M. Jones, Jr. and K. R. Rettig, J. Am. Chem. Soc. 1965, 87, 4013; M., Jr., Jones and K. R. Rettig, J. Am. Chem. Soc. 1965, 87, 4015.
83.(a) J. Weber and U. H. Brinker, Angew. Chem. Int. Ed. Engl. 1997, 36, 1623. (b) R. A. Wagner and U. H. Brinker, Chem. Lett. 2000, 3, 246. See also N. C. Yang and T. A. Marolewski, J. Am. Chem. Soc. 1968, 90, 5644, who first proposed this mechanism.
84.(a) J. E. Jackson, G. B. Mock, M. L. Tetef, G.-x. Zheng, and M. Jones, Jr., Tetrahedron 1985, 41, 1453; (b) J. Weber, L. Xu, and U. H. Brinker, Tetrahedron Lett. 1992, 33, 4537.
85.J. M. Cummins, T. A. Porter, and M. Jones, Jr., J. Am. Chem. Soc. 1998, 120, 6473; J. M. Cummins, I. Pelczer, and M. Jones, Jr., J. Am. Chem. Soc. 1999, 121, 7595.
86.Singlet carbene O H reactions have been very thoroughly reviewed by Kirmse: W. Kirmse, in Ref. 10, pp.1ff.; W. Kirmse, in Ref. 12, pp. 1ff.
87.Excellent reviews of early research on C H insertion reactions of carbenes may be found in Refs. 5 and 6.
88.W. von E. Doering and H. Prinzbach, Tetrahedron 1959, 6, 24.
89.W. Kirmse and M. Buschoff, Chem. Ber. 1969, 102, 1098.
90.G. L. Closs and L. E. Closs, J. Am. Chem. Soc. 1969, 91, 4549; G. L. Closs and A. D. Trifunac, J. Am. Chem. Soc. 1969, 91, 4554.
91.H. D. Roth, J. Am. Chem. Soc. 1972, 94, 1761.
92.I. Tabushi, Z.-i. Yoshida, and N. Takahashi, J. Am. Chem. Soc. 1970, 92, 6670.
93.K. Steinbeck, Chem. Ber. 1979, 112, 2402; K. Steinbeck and J. Klein, J. Chem. Res. (S)
1980, 94.
94.K. Steinbeck and J. Klein, J. Chem. Res. (S) 1978, 396.
95.D. Seyferth, V. M. Mai, and M. E. Gordon, J. Org. Chem. 1970, 35, 1993.
96.D. Seyferth, J. M. Burlitch, K. Yamamoto, S. S. Washburne, and C. J. Attridge, J. Org. Chem. 1970, 35, 1989.
97.D. Seyferth and Y. M. Cheng, J. Am. Chem. Soc. 1973, 95, 6763.
98.R. A. Pascal, Jr. and S. Mischke, J. Org. Chem. 1991, 56, 6954.
99.H. M. L. Davies, Q. Jin, P. Ren, and A. Yu. Kovalevsky, J. Org. Chem. 2002, 67, 4165.
100.D. Seyferth, R. Damrauer, J. Y.-P. Mui, and T. F. Jula, J. Am. Chem. Soc. 1968, 90, 2944.
101.H. D. Roth, J. Am. Chem. Soc. 1971, 93, 1527.
102.I. R. Likhotvorik, K. Yuan, D. W. Brown, P. A. Krasutsky, N. Smyth, and M. Jones, Jr.,
Tetrahedron Lett. 1992, 33, 911.
103.L. Xu, W. B. Smith, and U. H. Brinker, J. Am. Chem. Soc. 1992, 114, 783. This reference contains an excellent bibliography of dihalocarbene C H insertion reactions.
104.M. P. Doyle, J. Taunton, S.-M. Oon, M. T. H. Liu, N. Soundararajan, M. S. Platz, and J. E. Jackson, Tetrahedron Lett. 1988, 29, 5863.
105.J. E. Jackson, N. Soundararajan, M. S. Platz, M. P. Doyle, and M. T. H. Liu, Tetrahedron Lett. 1989, 30, 1335.
326SINGLET CARBENES
106.R. A. Moss and S. Yan, Tetrahedron Lett. 1998, 39, 9381.
107.R. A. Moss and S. Yan, Org. Lett. 1999, 1, 819.
108.See Ref. 5b, pp. 236 ff.
109.W. J. Baron, M. R. De Camp, M. E. Hendrick, M. Jones, Jr., R. H. Levin, and M. B. Sohn, in Ref. 6, pp. 19 ff.
110.See R. A. Moss, in Ref. 10, pp. 59 ff.
111.See R. Bonneau and M. T. H. Liu, in Ref. 11, pp. 1 ff.
112.See M. Jones, Jr., in Ref. 11, pp. 77 ff.
113.See M. S. Platz, in Ref. 11, pp. 133 ff.
114.See D. C. Merrer and R. A. Moss, in Ref. 12, pp. 53 ff. The Appendix to this reference contains extensive tables of rates and activation parameters for the intramolecular insertions of many alkylcarbenes.
115.See G. Szeimies, in Ref. 12, pp. 269 ff.
116.(a) R. Bonneau and M. T. H. Liu, J. Am. Chem. Soc. 1989, 111, 5973. (b) E. J. Dix, M. S. Herman, and J. L. Goodman, J. Am. Chem. Soc. 1993, 115, 10424; J. A. LaVilla and
J.L. Goodman, J. Am. Chem. Soc. 1989, 111, 6877.
117.J. W. Storer and K. N. Houk, J. Am. Chem. Soc. 1993, 115, 10426.
118.A. E. Keating, M. A. Garcia-Garibay, and K. N. Houk, J. Phys. Chem. A 1998, 102, 8467.
119.T. V. Albu, B. J. Lynch, D. G. Truhlar, A. C. Goren, D. A. Hrovat, W. T. Borden, and
R.A. Moss, J. Phys. Chem. A 2002, 106, 5323.
120.S. Celebi, S. Levya, D. A. Modarelli, and M. S. Platz, J. Am. Chem. Soc. 1993, 115, 8613;
R.Bonneau, M. T. H. Liu, and M. T. Rayez, J. Am. Chem. Soc. 1989, 111, 5893.
121.M. T. H. Liu and R. Bonneau, J. Am. Chem. Soc. 1996, 118, 8098.
122.A. Nickon, Acc. Chem. Res. 1993, 26, 84.
123.J. D. Evenseck and K. N. Houk, J. Phys. Chem. 1990, 94, 5518.
124.M. H. Sugiyama, S. Celebi, and M. S. Platz, J. Am. Chem. Soc. 1992, 114, 966.
125.M. T. H. Liu, Acc. Chem. Res. 1994, 27, 287.
126.D. C. Merrer, R. A. Moss, M. T. H. Liu, J. T. Banks, and K. U. Ingold, J. Org. Chem. 1998, 63, 3010.
127.(a) M. T. H. Liu and R. Bonneau, J. Am. Chem. Soc. 1990, 112, 3915; (b) R. Bonneau,
M.T. H. Liu, and M. T. Rayez, J. Am. Chem. Soc. 1989, 111, 5973.
128.M. T. H. Liu, R. Bonneau, S. Wierlacher, and W. Sander, J. Photochem. Photobiol., A: Chem. 1994, 84, 133.
129.S. Wierlacher, W. Sander, and M. T. H. Liu, J. Am. Chem. Soc. 1993, 115, 8943.
130.M. T. H. Liu and R. Bonneau, J. Phys. Chem. 1989, 93, 7298.
131.R. A. Moss, G. J. Ho, S. Shen, and K. Krogh-Jespersen, J. Am. Chem. Soc. 1990, 112, 1638. See also G.-J. Ho, K. Krogh-Jespersen, R. A. Moss, S. Shen, R. S. Sheridan, and
R.Subramanian, J. Am. Chem. Soc. 1989, 111, 6875.
132.R. A. Moss, G.-J. Ho, and W. Liu, J. Am. Chem. Soc. 1992, 114, 959.
133.B. T. Hill, Z. Zhu, A. Boeder, C. M. Hadad, and M. S. Platz, J. Phys. Chem. A 2002, 106, 4970.
134.(a) J. P. Pezacki, P. Couture, J. A. Dunn, J. Warkentin, P. D. Wood, J. Lusztyk, F. Ford, and M. S. Platz, J. Org. Chem. 1999, 64, 4456; (b) D. A. Modarelli, S. Morgan, and M. S. Platz, J. Am. Chem. Soc. 1992, 114, 7034.
REFERENCES 327
135.I. R. Likhotvorik, E. Tippmann, and M. S. Platz, Tetrahedron Lett. 2001, 42, 3049.
136.(a) Z. Zhu, T. Bally, L. L. Stracener, and R. J. McMahon, J. Am. Chem. Soc. 1999, 121, 2863; Y. Wang, T. Yuzawa, H. Hamaguchi, and J. P. Toscano, J. Am. Chem. Soc. 1999, 121, 2875; J.-L. Wang, I. Likhotvorik, and M. S. Platz, J. Am. Chem. Soc. 1999, 121, 2883; (b) F. Kaplan and G. K. Meloy, J. Am. Chem. Soc. 1966, 88, 950.
137.(a) H. M. Frey, Pure Appl. Chem. 1964, 9, 527. (b) A. M. Mansoor and I. D. R. Stevens, Tetrahedron Lett. 1966, 1733; (c) K.-T. Chang and H. Shechter, J. Am. Chem. Soc. 1979, 101, 5082.
138.M. S. Platz, in Ref. 11, p. 133ff.
139.J. M. Fox, J. E. Gillen Sacheri, K. G. L. Jones, M. Jones, Jr., Jones, P. B. Shevlin,
B.Armstrong, and R. Sztyrbicka, Tetrahedron Lett. 1992, 33, 5021.
140.G. Wu, M. Jones, Jr., W. von E. Doering, and L. H. Knox, Tetrahedron, 1997, 53, 9913.
141.(a) H. Glick, I. R. Likhotvorik, and M. Jones, Jr., Tetrahedron Lett. 1995, 36, 5715;
(b) B. M. Armstrong, M. L. McKee, and P. B. Shevlin, J. Am. Chem. Soc. 1995, 117, 3689.
142.W. Kirmse, B. G. von Bulow, and H. Schepp, Justus Liebigs Ann. Chem. 1966, 41, 691.
143.H. Huang and M. S. Platz, J. Am. Chem. Soc. 1998, 120, 5990.
144.R. A. Moss and N. J. Turro, Ref. 9, p. 213ff.
145.M. S. Platz, Ref. 11, p. 164.
146.M. Nigam, M. S. Platz, B. Showalter, J. Toscano, R. Johnson, S. C. Abbot, and M. M. Kirchhoff, J. Am. Chem. Soc. 1998, 120, 8055.
147.(a) P. E. Eaton and K. L. Hoffman, J. Am. Chem. Soc. 1987, 109, 5285; (b) P. E. Eaton and
R.B. Appell, J. Am. Chem. Soc. 1990, 112, 4055.
148.W. R. White, M. S. Platz, N. Chen, and M. Jones, Jr., J. Am. Chem. Soc. 1991, 113, 4981;
D.A. Hrovat and W. T. Borden, J. Am. Chem. Soc. 1996, 118, 1535.
149.See also R. T. Ruck and M. Jones, Jr., Tetrahedron Lett. 1998, 39, 4433.
150.R. T. Ruck and M. Jones, Jr., Tetrahedron Lett. 1998, 39, 2277.
151.M. I. Kahn and J. Goodman, J. Am. Chem. Soc. 1995, 117, 6635.
152.R. A. Moss, S. Yan, and K. Krogh-Jespersen, J. Am. Chem. Soc. 1998, 120, 1088;
K.Krogh-Jespersen, S. Yan, and R. A. Moss, J. Am. Chem. Soc. 1999, 121, 6269.
153.D. S. Wulfman, B. Poling, and R. S. McDaniel, Jr., Tetrahedron Lett. 1975, 4519.
154.J. P. Toscano, Ref. 11, p. 231ff.
155.See Reference 79.
156.R. C. Joines, A. B. Turner, and W. M. Jones, J. Am. Chem. Soc. 1969, 91, 7754.
157.H. Staudinger and R. Endle, Chem. Ber. 1913, 46, 1437; F. O. Rice and J. D. Michaelsen,
J.Phys. Chem. 1962, 66, 1535.
158.J. A. Myers, R. C. Joines, and W. M. Jones, J. Am. Chem. Soc. 1970, 92, 4740.
159.G. G. Vander Stouw, A. R. Kraska, and H. Shechter, J. Am. Chem. Soc. 1972, 94, 1655.
160.W. J. Baron, M. Jones, Jr., and P. P. Gaspar, J. Am. Chem. Soc. 1970, 92, 4739.
161.For a nice review of the early work, and provocative mechanistic suggestions, see: P. P. Gaspar, J.-P. Hsu, S. Chari, and M. Jones, Jr., Tetrahedron, 1985, 41, 1479.
162.O. L. Chapman, J. W. Johnson, R. J. McMahon, and P. R. West, J. Am. Chem. Soc. 1988, 110, 501.
328SINGLET CARBENES
163.P. R. West, O. L. Chapman, and J.-P. LeRoux, J. Am. Chem. Soc. 1982, 104, 1779; R. J. McMahon, C. J. Abelt, O. L. Chapman, J. W. Johnson, C. L. Kreil, J.-P. LeRoux, M. Mooring, and P. R. West, J. Am. Chem. Soc. 1987, 109, 2459; O. L. Chapman and C. J. Abelt, J. Org. Chem. 1987, 52, 121.
164.R. Warmuth, Eur. J. Org. Chem. 2001, 423.
165.R. Warmuth and M. A. Marvel, Chem. Eur. J. 2001, 7, 1209.
166.P. C. Venneri and J. Warkentin, J. Am. Chem. Soc. 1998, 120, 11182.
167.(a) N. Merkley, M. El-Saidi, and J. Warkentin, Can. J. Chem. 2000, 78, 356. (b) N. Merkley and J. Warkentin, Can. J. Chem. 2000, 78, 942.
168.R. A. Moss, B. K. Wilk, and L. M. Hadel, Tetrahedron Lett. 1987, 28, 1969.
169.R. A. Moss, Acc. Chem. Res. 1999, 32, 969.
170.R. A. Moss, L. A. Johnson, S. Yan, J. P. Toscano, and B. M. Showalter, J. Am. Chem. Soc. 2000, 122, 11256.
171.S. Yan, R. R. Sauers, and R. A. Moss, Org. Lett. 1999, 1, 1603.
172.M. A. Kesselmayer and R. S. Sheridan, J. Am. Chem. Soc. 1986, 108, 99, 844.
173.R. A. Moss, Y. Ma, F. Zheng, R. R. Sauers, T. Bally, A. Maltsev, J. P. Toscano, and B. M. Showalter, J. Phys. Chem. A, 2002, 106, 12280.
174.R. A. Moss, F. Zheng, R. R. Sauers, and J. P. Toscano, J. Am. Chem. Soc. 2001, 123, 8109.
175.R. C. Bingham and P. v. R. Schleyer, J. Am. Chem. Soc. 1971, 93, 3189.
176.R. A. Moss, F. Zheng, J.-M. Fede´, Y. Ma, R. R. Sauers, J. P. Toscano, and B. M. Showalter, J. Am. Chem. Soc. 2002, 124, 5258.

 CHAPTER 8
CHAPTER 8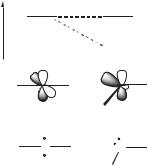
330 STABLE SINGLET CARBENES
tetravalency of carbon was not established, and therefore the existence of stable carbenes was considered to be quite reasonable. At the very beginning of the twentieth century, Staudinger and Kupfer demonstrated that carbenes, generated from diazo compounds or ketenes, were highly reactive species.2 It quickly became clear that their six valence electron shell, which defied the octet rule, was responsible for their fugacity. As a consequence, the quest for stable carbenes became an unreasonable target, and indeed remained so for quite some time! In the 1950s, Breslow3 and Wanzlick,4 realized that the stability of a carbene could be dramatically enhanced by the presence of amino substituents, but they were not able to isolate a ‘‘monomeric’’ carbene. It was only in 1988 that our group reported the synthesis of a stable carbene, namely, a (phosphino)(silyl)carbene.5
Since this discovery, a few persistent triplet carbenes6 have been prepared and other types of stable singlet carbenes have been isolated. Triplet carbenes are discussed in Chapter 9 by H. Tomioka in this volume, and therefore this chapter will be focused on singlet carbenes.
2. SINGLET VERSUS TRIPLET GROUND STATE?
The divalent carbon atom of carbenes has only six electrons in its valence shell. Four are used to make s bonds with the two substituents, and therefore two nonbonding electrons remain. To understand, the difference between the singlet and the triplet state, let us consider a prototypal carbene. The carbon atom can be either linear or bent, each geometry is describable by a specific hybridization. The linear geometry implies an sp hybridized carbene center with two nonbonding degenerate orbitals ( px and py). Bending the molecule breaks this degeneracy and the carbon atom adopts an sp2-type hybridization: the py orbital remains almost unchanged (it is usually called pp), while the orbital that starts as a pure px orbital is stabilized because it acquires some s character (it is therefore called s) (Fig. 8.1). The carbene ground-state multiplicity is related to the relative energy of the s and pp orbitals. The singlet ground state is favored by a large s-pp separation; Hoffmann assumed
E |
py |
pπ |
|
px |
|
|
|
σ |
|
py |
p |
|
px |
π |
|
|
σ |
linear bent
CC
Figure 8.1. Relationship between the carbene bond angle and the nature of the frontier orbitals.