

Molecular Heterogeneous Catalysis, Wiley (2006), 352729662X
.pdf316 Chapter 7
Figure 7.1. Model of the hexakinase enzyme: (a) the free cavity in hexakinase, (b) adsorbed glucose in the cavity[2c].
Figure 7.1c. Model of the hexakinase enzyme: adsorption of glucose[2a].
influence both its intrinsic electronic structure and its reaction environment, both of which will a ect its catalytic performance. This will be described in the next section.
We will give a detailed description of recent insights obtained especially for the widely studied hexakinase enzyme, which catalyzes the phosphorylation of glucose. The phosphorylation of glucose by hexakinase has been modeled in detail by simulating the reaction events that can occur in the enzyme cleft[2]. The free hexakinase enzyme molecule is
Mechanisms in Biocatalysis; Relationship with Chemocatalysis 317
shown in Fig. 7.1a. The glucose molecule has not yet been attached. Its shape is significantly altered upon the adsorption of glucose, as seen in Fig. 7.1b. The reaction does not appear to follow the lock and key model[3], which was originally proposed to explain the stereoselectivity in enzymes. The lock and key model would suggest that glucose would exactly match the open cleft in the hexakinase enzyme. Instead, the enzyme cavity shape and hence overall three-dimensional structure adapt to accommodate optimally the glucose substrate molecule. This is consistent with the “induced fit model” proposed by Koshland[4]. In this model, small induced changes within the reaction complex lower the activation energy of reaction. The cavity shape adapts leading to an optimum fit between the reactive groups in the adjusted cavity and the reactant. Such changes cannot occur in the rigid inorganic frameworks of heterogeneous catalysts such as zeolites discussed earlier.
Upon the adsorption of the glucose molecule into the enzyme cavity, the cavity closes as observed in Fig. 7.1b. The cavity continues to change as the reaction proceeds, which helps to drive the reaction over the potential energy surface to the product state. Desorption of the product molecules requires reopening of the cavity in order to release them. This process can be aided by the coadsorption of an additional reactant molecule at a second peptide binding site (allosteric e ect). This reduces the interaction between product molecules and cavity, assists desorption and decreases the tendency of the enzyme to become deactivated by product poisoning.
A unique feature of the enzyme is multi-point bonding of reaction intermediates to the enzyme cavity and the participation of several protein substituents often belonging to di erent amino acids in the activation of a substrate (Fig. 7.1c).
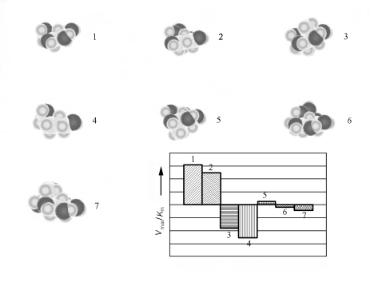
The optimum induced fit between the enzyme cavity and the reacting substrate enables the enzyme for a particular reaction to discriminate readily between reactants that di er in size. This is illustrated in Fig. 7.2 for the conversion of glucose and closely related molecules by the hexakinase enzyme.
A good example of this is found in the studies by Rose[6] on the conversion of malate to fumarate conversion. He showed for the enzyme fumarase that malate combines with a conformation of the enzyme that accommodates malate more easily than fumarate. As the chemical transition proceeds the cavity changes shape and provides for a better accommodation of the fumarate. For net catalysis to proceed, the fumarase must finally make a transition to the malate-preferring confirmation. In this step fumarate desorbs. The minimum scheme for catalysis by an enzyme is than as follows:
E0 + S ←→ Es S ←→ EP.T ←→ EpP ←→ E1TP
where E0 is an enzyme form that has a better fit to substrate S than product P. E0 changes its conformation to Es when S adsorbs. When reaction proceeds, a pre transition (PT) intermediate is formed and enzyme conformation adapts. This state continues towards the formation of Ep which stabilizes the formation of the product just enough for the product P to form but not so far that it will not desorb. After reaction, the state of the enzyme E1T has to return to its original state, E0.
Optimization of reaction complex topology implies the positioning of charged enzyme groups such that there is an optimum interaction with (activated) reactants. Significant protonation (in protolytic enzymes) or electron transfer (in redox catalysis) can already occur in the adsorbed state. This is due to the unique electrostatic properties of the enzyme, which is shielded from the solvent by its hydrophobic peptide framework.
318 Chapter 7
Figure 7.2. The rate of conversion of glucose related molecules by hexakinase. Dependence on molecule shape[5]. The molecules converted are labeled with a number.
The formation of the optimum pre-transition-state complex requires some energy since the enzyme restructures and the state of the substrate molecules becomes slightly deactivated compared to its most stable adsorption state. The activation energy with respect to the pretransition state may be only a few kJ/mol. For a hydrolysis reaction, the enzyme typically lowers the activation barriers by 30–50 kJ/mol compared with the acidor basecatalyzed reaction in solution. This overall lowering of the apparent activation energy is the result of the optimized electrostatic interaction and hydrogen bonding that occurs in the initial reaction complex. The energy is only partially o -set by the cost in energy of restructuring of the enzyme peptide framework. The interaction between substrate and the atoms of the catalytic reactive center tends to weaken bonds within the catalytic center itself, thus initiating its relaxation and restructuring. As we have discussed previously for chemo catalysis, reaction energies not only depend on the changes in bond energies that occur within the substrate or between substrate and active site in the catalyst, but also within the catalyst itself.
The concept of a pretransition-state stabilization has also been extensively discussed in the chapter on zeolite catalysis (Chapter 4). In addition, the stereochemical selectivity found in chemocatalytic systems is often due to stabilization of reagents in a particular conformation before actual bond activation occurs.
Enzyme catalysts typically have low activation energies. The synchronized action of multi-atom displacements to optimize the interaction energy with the pretransition-state complex implies a decrease in the entropic state of the complex. This has been analyzed by Liktenstein[7], who formulated the principle of “optimum motion”, in contrast to the principle of “minimum motion”.
According to the latter, the less that nuclei have to change their position in the transition of an elementary reaction step, the lower is the reorganization energy required. This lowers both the overall energy and the activation energy for the reaction. The concerted

Mechanisms in Biocatalysis; Relationship with Chemocatalysis 319
motion of many atoms will stabilize the pretransition-state complex, thus leading to a small activation energy as the result. According to the principle of optimum motion, the number of nuclei whose configuration is changed in an elementary reaction step must be su ciently large to lower the activation energy with respect to the adsorbed state. At the same time, this number has to be su ciently small so that the synchronization probability along the reaction coordinate is not too low.
Liktenstein proposed a condition that has to be satisfied so that ksyn is faster than the reaction rate of the direct reaction kdir:
ksyn |
= αsynexp |
Edir − Esyn |
> 1 |
(7.2) |
|
kT |
|||
kdir |
|
|
||
where αsyn is the synchronization factor, which is the ratio of the preexponential factors of the synchronous and direct processes
αsyn = |
2n−1 |
πEsyn |
n−1 |
(7.2a) |
||
ϕcr > ϕ0; Esyn > nkT |
||||||
|
n |
|
nkT |
|
|
|
αsyn = |
n |
|
|
|
ϕcr < ϕ0; Esyn > nkT |
(7.2b) |
|
|
|
|
|||
2n−1 |
|
|
|
|||
where ϕ is a parameter that denotes the displacement of the nuclei, ϕcr is some critical value of this displacement and n is the number of vibrational degrees of freedom of the nuclei participating in the concerted transition.
For activation energies typical for enzymatic reactions, cooperation of each new nucleus in the transition state can lead to a ten-fold decrease in the reaction rate. Liktenstein used the Eq. (7.2)to analyze di erent mechanistic schemes proposed for an enzymatic reaction. One concludes that reaction paths involving a smaller number of synchronizing atoms, but proceeding through covalently bonded intermediates, as we discussed for chymotrypsin (Section 4.4, p. 203), are usually preferred.
When a reactant molecule adsorbs on a particular site, entropy is lost compared with the reactant state in solvent or gas phase. This was described earlier in the chapter on zeolites. Within the rigid lock and key model, this entropy loss would be maximum, thus reducing the free energy gained upon adsorption. This is an additional reason why an optimum fit between reactant and enzyme cavity is not preferred. When the fit between the reactant and the cavity is not optimum, the reactant will maintain some mobility in the adsorbed state, hence the entropy loss is less. The basic mechanistic principles for enzyme catalysis discussed so far include the induced fit of the enzyme cavity as a response to substrate shape and size, pretransition-state stabilization of activated molecules and the principle of optimum motion. A reaction that proceeds through intermediates via transient covalent bonds is preferred.
Enzyme catalysis, as discussed so far, occurs at a single site of the protein. Enzymes that modify proteins or macromolecules have to distinguish between similar substrate sites within larger dissimilar molecules. Discrimination between di erent substrates bearing the
320 Chapter 7
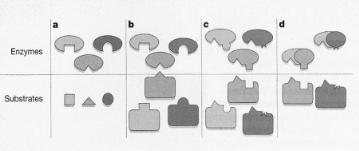
Figure 7.3. How enzymes select their substrates. (a, b), In general, enzymes recognize their targets through complementary structural features between the substrate and the enzyme’s active site (indicated here by the shape of the “pocket”). Small substrates (a) and relatively small modification sites on proteins
(b) can be recognized by this mechanism. (c) Some enzymes make additional, specific contacts with the substrate that enable them to distinguish between proteins that have identical or related sites of modification. (d) Cyclin-dependent protein kinases (CDKs) relegate that function to the exchangeable cyclin subunit, enabling a single CDK catalytic subunit to exist in numerous forms with di erent specificities. Adapted from Wittenberg, Nature 434, 35 (2005).
same motif occurs through additional specific interactions between sites other than the reaction center of the enzyme and substrate. This is illustrated in Fig. 7.3.
Such cooperativity has, for instance, been shown for cell cycle-regulating cyclin-depen- dent kinases (CDK) or baker’s yeast. These CDKs consist of the combination of a subunit cyclin and a catalytic subunit. The presence of a particular hydrophobic structural motif on cyclin in combination with the catalytic subunit imports unique reactivity with respect to the phosphorylation of particular substrates with di erent size.
7.3 ATP-Synthase Mechanism; a Rotating Carousel with Multiple Catalytic Sites
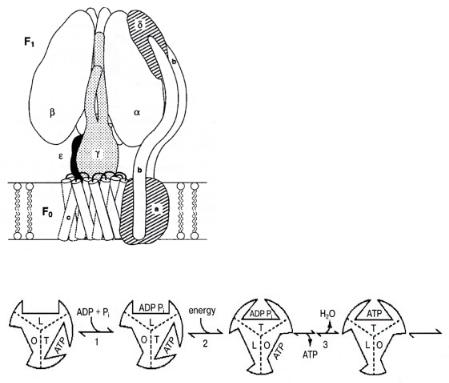
The enzyme that catalyzes the hydrolysis or synthesis of adenosine triphosphate (ATP) demonstrates a unique feature in which a rotational motion of the protein that contains at least three reaction centers is coupled with di erent stages of the catalytic reaction occurring at each of these centers. The enzyme protein is attached to a proton translocating membrane. The energy changes due to ATP formation or hydrolysis is coupled with proton transport through the membrane, which is important to the biosystem’s energy housekeeping. The rotary motion of the ATP synthase enzyme couples the chemical potential changes of the chemical reaction with chemical potential changes over the membrane. We will follow closely two important review papers[8,9] that help to elucidate the mechanism of catalytic action of this catalyst. A model representation of the F1 enzyme protein attached to the membrane F0 is given in Fig. 7.4.
F1 is a globular aggregate attached through subunit γ to F0, the membrane protein complex. γ is the shaft around which three αβ subunits rotate. Proton transfer occurs through c and a channels. The three catalytic sites are located at α/β interfaces. Sequential opening and closing of the catalytic sites drives rotation of the γ subunit. Conformational signals travel from one catalytic site to another. ATP synthesized at a high-a nity catalytic site will desorb using energy produced by proton transfer through F0. The F1 unit then rotates by 120◦. The original three-site model proposed by Cross[9] illustrating the sequence of reaction steps that occur is shown in Fig. 7.5.
Mechanisms in Biocatalysis; Relationship with Chemocatalysis 321
Figure 7.4. A model of the E. coli F0F1 ATP-synthase, after Ogilvie et al.[10].
Figure 7.5. A three-site model for the binding change mechanism. Adapted from R.L. Cross[9].
Tight(T), loose(L) and open(O) phases of three catalytic centers are proposed. ATP will only be released from F1 once ATP is formed at a di erent site by adsorption of ADP and phosphate. The conformational changes that occur in the enzyme cavities when the reaction proceeds are communicated through the subunits to the other sites. This allotropic communication implies synchronization of the di erent phases of the reaction at the di erent reaction centers.
The coupling of proton transfer through a membrane to the generation of rotary motion is known from other biochemical studies. For instance, it is also responsible for the flagellar rotation of swimming bacteria (see ref. [1]). A rotational velocity of 100 revolutions per second, consistent with the time scale of catalytic reactions, is typically reached. Each revolution is driven by a flow of 1000 protons across the membrane[11]. Rotary force is generated by the interplay of receptors on the rotating ring and the half-closed proton channels on the membrane.
The H+ translocates through the membrane in three steps. First it adsorbs into a half channel of the membrane, then it connects with a proton acceptor site on the rotating F1 ring. In a rotation, the proton is back-donated to the empty other half-channel of the membrane, that transfers the proton to the other side of the membrane. Rotational motion will only occur when one half-channel is unoccupied by a proton and the other half is empty.
322 Chapter 7
7.4 Carbonic Anhydrase
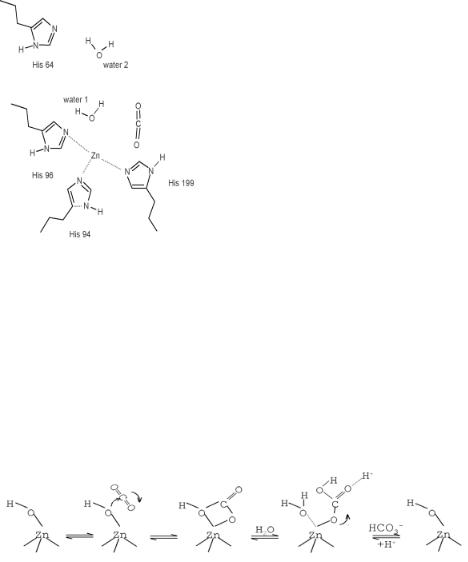
As a first comparison of a chemocatalytic reaction with an analogous enzyme-catalyzed reaction, we discuss the hydrolysis of CO2 by H2O to give HCO3 − by the enzyme carbonic anhydrase. The reaction steps involved in the enzyme catalyzed mechanism will be compared with the chemocatalytic steps involved in the hydrolysis of acetonitrile by a Zn2+ containing zeolite as discussed on page 186 in Chapter 4. Similarly to the zeolite, the interior of the enzyme is hydrophobic except for the region close to the Zn2+ center. Its structure is shown in Fig. 7.6.
Figure 7.6. The catalytic center of carbonic anhydrase[12a].
The Zn2+ ion is attached to three basic nitrogen atoms of imidazole groups that are part of histidine peptides of the enzyme protien framework[12]. The positive charge of Zn2+ is compensated for by negatively charged glutamate residues in the second coordination shell of Zn2+. A proton-transfer reaction between the negatively charged glutamate groups and imidazole occurs upon proton attachment that is the result of water dissociation. Two or three H2O molecules are involved in the reaction[13]. One molecule dissociates on Zn2+ to give ZnOH+ and a proton that attaches to imidazole which is stabilized upon the transfer of another proton from imidazole to neighboring glutamate. Similarly as in the zeolite (see Fig. 4.23), the second water molecule facilitates the initial proton-transfer reaction. The proton is transferred through the second water molecule to a basic proton-accepting site.
Figure 7.7. The hydrolysis reaction of CO2 to bicarbonate[12b].
Mechanisms in Biocatalysis; Relationship with Chemocatalysis 323
The OH− bonded to Zn2+ reacts in a consecutive step with CO2 to form a bicarbonate ion via the sequence shown in Fig. 7.7. The bicabonate is released when another water molecule adsorbs and dissociate to form new OH− sites. The proton initially attached to the His64 peptide is then released through the water network to charge compensate the bicarbonate molecule. This closes the catalytic cycle. The reactive center is regenerated in the last adsorption-induced desorption step.
The uniqueness of the enzyme relates to the hydrophobic environment of Zn2+ and the protection of this site against deactivating reactions, for instance, leaching of Zn2+ by dissolution of Zn2+ into the water phase. The leaching of Zn2+ is a problem that tends to occur with the zeolite catalyst. In enzyme catalysis, bicarbonate is formed at neutral pH, which is very di erent from the basic conditions used in the non-catalytic system. In addition, the barrier for the dissociative adsorption of H2O to liberate H2CO3 is lowered in a unique way, by the stabilization of the protons that are formed through a sequence of hydrogen bonds they create with the surrounding basic imidazole groups. Such a unique optimized environment is absent in the case of the zeolite.
The similarities between the nitrile hydrolysis and CO2 hydrolysis system relate to the heterolytic H2O-assisted splitting of H2O and the importance of the adsorption-induced desorption of the reactant molecule. The primary di erence between the enzyme and the zeolite relates to the specific interactions with the imidizole groups in the histidine framework, which can simultaniously interact with the reaction center to aid bond cleavage and bond formation reactions.
7.5 Biomimicking of Enzyme Catalysis
The development of chemocatalytic materials that mimic transformations carried out in enzyme catalysis has been a major goal of molecular imprinting approaches[14] and more generally biomimetic methods. The copolymerization of decomposable organic complexes into an inorganic framework should lead to the formation of cavities that have a prearranged shape that can be produced via the decomposition of a templating complex. The cavity then contains activating groups that are arranged such that a particular transitionstate structure is stabilized.
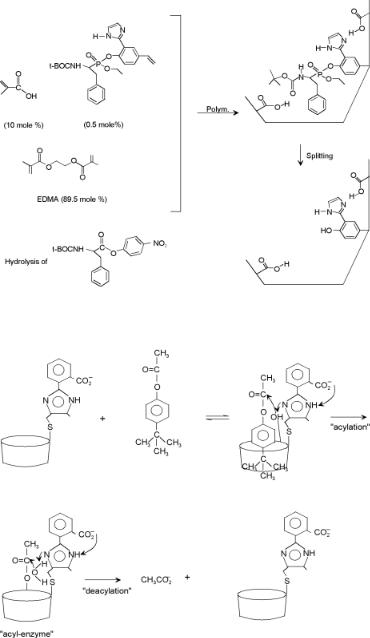
In Chapter 4, we briefly discussed the hydrolysis of a peptide bond (Fig. 4.23a) by chymotrypsin, through the intermediate formation of a tetrahedral transition state. Figure 7.8 illustrates an approach to mimic such a reaction center for the hydrolysis of an ester using a polymer matrix. The reaction center is created using a templating phosphatecontaining molecule that becomes part of the framework mimicking the transition state of the molecule to be hydrolyzed. The catalyst cavity is subsequently created by removal of the phosphate-containing molecular fragment before catalysis. In Chapter 8, self-organizing biocatalytic systems exploiting the immuno system will be discussed in which the reaction centers are generated by triggering the system using molecules which have shapes similar to those of the transition-state complexes for the reactions to be catalyzed. In addition directed synthesis approaches can also be used.
D’Souza and Bender[15] proposed the synthesis of a hydrolyzing cavity that mimicks chymotrypsin by attachment of selected substituents to a dextrine cavity (Fig. 7.9).
The two examples shown in Figs. 7.9 and 7.10 illustrate attempts at synthesizing a biomimetic flexible environment with well-controlled electrostatics strategically placed in a small cavity to provide concerted activation of probe molecules.
324 Chapter 7
Figure 7.8. Schematic representation of the preparation of a catalyst by a transition-tate analogue using labile covalent binding and noncovalent binding[14].
Figure 7.9. The mechanism of action of artificial chymotrypsin[15].
The principles of supramolecular catalysis relate to enzyme catalysis, because weak chemical bonds involving well defined hydrogen bonds are essential. Lehn[16] formulated two main steps required for supramolecular reactivity and catalysis:
Mechanisms in Biocatalysis; Relationship with Chemocatalysis 325
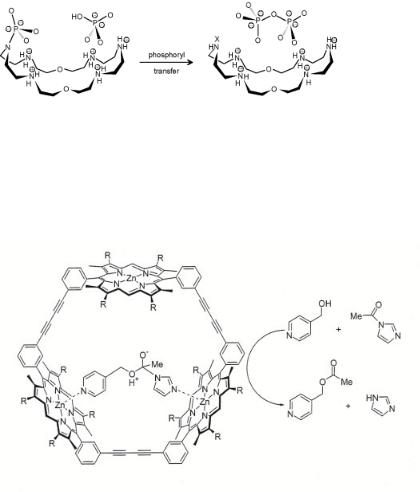
Figure 7.10. Cocatalysis: pyrophosphate synthesis by phosphoryl transfer[17].
–Binding which selects the substrate. Selectivity requires molecular recognition along with the optimization of the pretransition-state configuration.
–Transformation of the bound species into products. The supramolecular system has to transform reactants selectively to products that are accommodated in the supramole– cule structure, but also readily desorb.
Figure 7.11. Tetrahedral intermediate of the acyl-transfer reaction bound inside the cavity of a porphyrin trimer. Adapted from V.F. Slagt [Thesis, Amsterdam (2004)].
The D’Souza and Bender system shows many of these features. Systems that induce bond formation require the presence of several binding and reactive groups. The catalytic molecule should act as a co-receptor bringing together reactant, substrate and intermediate complex. As an example, Hisselni and Lehn[17] demonstrated pyrophosphate formation from the intermediate phosphoramidate formed by phosphorylation of the macrocycle by ATP. In a second reaction step, the phosphoramidate reacts with a phosphate group to form pyrophosphate (see Fig. 7.10).
Enzyme-mimicking systems that contain metal cations have also been designed. A very elegant supramolecular assembly was designed by Sanders et al.[18] (see Fig. 7.11). They constructed trimeric porphyrin structures where Zn2+ porphyrin moieties function as templates for the organization of substrates into a conformationally optimal configuration that undergoes an e cient acyl-transfer reaction or that lead to Diels–Alder products.