

Molecular Heterogeneous Catalysis, Wiley (2006), 352729662X
.pdf296 Chapter 6
NOads + NOads −→ N2O + Oads |
(6.27) |
NOads + Nads −→ N2O |
(6.28) |
Theoretical studies on Cu[51] and Pt[52] have shown that the associative mechanism is the preferred reaction (see Table 6.1 also) As indicated in the upper portion of Fig. 6.21, NO can in principle also react with NH3 to form N2. In ammonia oxidation catalysis, this is known as the Fogel mechanism. The reaction of NO and NH3 in the presence of oxygen is a well-known reaction used commercially at higher temperatures to remove NO from exhaust emission streams by its injection with ammonia as a sacrificial reductant. The catalysts that are typically used to carry out selective catalytic reduction (SCR) are based on MoO3, V2O5, WO3 and Cr2O3 (see Chapter 5, Section 5.6.10). Although these catalysts are less reactive than Pt, they o er much higher selectivities at higher temperatures.
Figure 6.22. Ammonia oxidation activation energies and overall reaction energies on Cu(111)[53].
Figure 6.22 highlights the reaction energies and the activation energies for the oxidation of ammonia over the Cu(111) surface. Note the exothermicity of the reactions that proceed directly via reaction with coadsorbed molecular oxygen.The experimental selectivity for the ammonia conversion reaction as a function of temperature[54] over a Cu catalyst is shown in Figure 6.23.
Figure 6.23. Ammonia oxidation on Cu/Al2 O3. Reaction conditions: NH3 = 1000 ppm, O2 = 10%, GHSV = 90,000[54].
Mechanisms for Aqueous Phase Heterogeneous Catalysis and Electrocatalysis 297
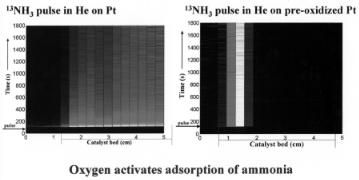
Figure 6.24. Adsorption of ammonia on Pt and pre-oxidized Pt sponge at 323 K. Pre-oxidized Pt: treated with 3% of O2 in a helium flow at 473 K for 1 hour[54b].
At low temperatures, the only products that form are N2O and N2. In situ spectroscopic studies of working Cu and Ag catalysts show that apart from adsorbed oxygen, there is a high surface coverage of nitrite and nitrate species[55] . Hence, on these metals at low temperature, N2 and N2O production is likely the result of consecutive reactions of NOx, the most abundant reaction intermediate (MARI), with NH3. N2 is formed by the reaction of nitrite with NH3, whereas N2O can also form via reaction of nitrate with ammonia (see also Section 6.4.1).
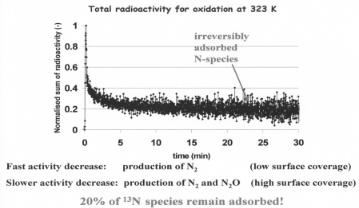
The low-temperature activation of NH3 over a Pt sponge[54,57] illustrates the complexity of this reaction on platinum. On Pt under steady-state conditions, the surface is predominantly covered with NHx and OHx species[56] . This is quite di erent from the surface state on Cu and Ag. As shown in Fig. 6.24, only a Pt catalyst precovered with oxygen will activate NH3, and at low temperature the products remain adsorbed as NHx intermediates. The results in Fig. 6.24 on the left show little ammonia reaction from a pulse of NH3 in He on Pt. In the absence of oxygen, NH3 does not activate, therefore no NHx product species are formed. The 13NH3 pulse rapidly moves through the catalyst bed. The reactivity of an ammonia–oxygen mixture on the reduced Pt at 320 K is initially quite high but rapidly declines after only a few minutes (see Fig. 6.25). The initial product, N2, subsequently becomes replaced by N2O. The production of both species rapidly decreases with time[54b]. These features are consistent with recombination of N adatoms to form N2 and N and O adatoms to form NO through low activation energy reaction channels. These reactions tend to be favored at the step edge sites. Nitrogen will desorb, but NO does not desorb at low temperatures. Adsorbed NO will di use away from the step edges to the terraces and will only desorb once N2O has been formed. The initial period of high activity at low temperature becomes inhibited at longer times. The terraces become covered with a high concentration of non-reactive NHx species. Radiochemical experiments at low temperature show that 20% of reacted NH3 remains adsorbed on the catalyst (Fig. 6.25) as an irreversible adsorbed N species.
Interestingly, single crystal experiments on Pt executed at low pressures have never observed the presence of N2O as has been seen in experiments carried out at higher pressures on Pt sponges. This is indicative of very di erent surface conditions in the vacuum experiment to those in the atmospheric pressure experiments presented above. These differences are very likely the result of the di erent surface compositions and coverages that form in reactions carried out under vacuum conditions and those at atmospheric pressure.
298 Chapter 6
Figure 6.25. Radioactivity from adsorbed 13 NHx species , oxidation at 323 K after an initial 13NH3 pulse in an oxygen flow. 20% of 13 NH species remain adsorbed[54b].
Ab initio kinetic Monte Carlo simulations carried out by Neurock and co-workers[52] on Pt(100) surface, for example, show that under UHV conditions NO does not react to form N2O but as the pressure is increased to atmospheric conditions N2O becomes one of the major reaction products along with N2. In these simulations NO dissociates when a surface has a low coverage with NO. N2O is formed when the coverage with NO is such that the dissociation of NO becomes suppressed.
Single-crystal UHV experiments, however, demonstrated the unique finding that N2 can be formed at temperatures as low as 210 K[57] if the reactions are carried out on
Pt(100). The Pt(100) surface was found to be active, in that it contains uniquely active 4-fold sites that allow for the dissociation of NO at significantly lower barriers than other surfaces. Similarly, the barrier for the recombination of Nads adatoms to form N2 is also rather low. The unusually low activation energies reported here are due to the elimination of sites which poison as the result of their reactants and/or products due to the elimination of metal atom sharing in the transition state. As was discussed in previous chapters, NO activation over Pt(100) and also at step edges is due to the removal of lateral repulsive interactions that arise from metal atom sharing.
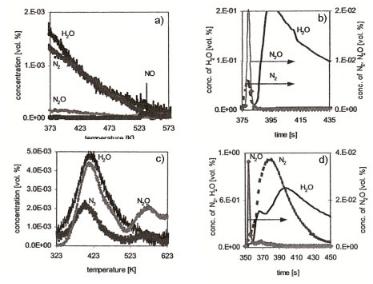
Additional experimental evidence on the reactivity of di erent surface intermediates initially formed on the Pt sponge has also been obtained via radiochemical temperatureprogrammed reaction experiments. These experiments probe the state of Pt and pre– oxidized Pt surfaces and their composition after exposure to a pulse of ammonia. A dramatically di erent reaction pattern is found as a function of temperature when in a subsequent experiment such a pretreated surface is exposed to a flow of H2, NO or NH3 [54] (see Fig. 6.26). The results from temperature-programmed desorption experiments show the desorption of a small amount of N2 after surface treatment with NH3 at 373 K and a small amount of NO desorption at 540 K (Fig. 6.26a). In contrast, experiments performed at fixed temperatures lead to the formation of much larger amounts of N2O and N2, which form immediately after H2 addition (Fig. 6.26b). This result is consistent with the proposal that in the presence of H2, the non-reactive surface oxygen (with respect to adsorbed NHx) is converted to surface hydroxyl groups which demonstrate a much higher reactivity for activating the NH bonds than atomic oxygen does. The formation of N2 and NO occurs from stepped or reactive surfaces as the (100) surfaces or step edges. The
Mechanisms for Aqueous Phase Heterogeneous Catalysis and Electrocatalysis 299
Figure 6.26. (a) A [13 N]NH3 pulse was adsorbed on the pre-oxidized platinum sponge kept under an He flow at 373 K followed after 170 s by temperature-programmed desorption. (b) A [13N]NH3 pulse was adsorbed on a pre-oxidized platinum sponge kept under an He flow followed after 380 s by a removal of N species with hydrogen: mass spectrum shown from the moment of hydrogen addition. (c) Formation of N2, N2O and H2 O measured by online mass spectrometry in temperature-programmed NO reaction after oxidation with ammonia at 323 K. (d) A [13 N]NH3 pulse was adsorbed on the pre-oxidized platinum sponge kept under an He flow followed after 350 s by a removal of N species with ammonia. Mass spectrum shown from the moment of NH3 addition.
reaction of NO responsible for low temperature N2O formation is
2NOads −→ N2O + Oads |
(6.29) |
Figure 6.26c shows the result of NO treatment of the Pt surface after exposure to ammonia and oxygen at low temperature. The products appear in two temperature regimes. The first product peaks appear at about 400 K and are the result of N2, N2O and H2O formation. A second set of peaks appear at 570 K, which correspond to the formation of N2O and N2. Reaction (6.29) is consistent with low-temperature N2O formation. The high-temperature N2O peak is consistent with a more di cult reaction sequence:
NOads + Nads −→ N2O |
(6.30) |
NO tends to desorb at the same higher temperature hence, surface vacancies become available for NO dissociation. Finally, Fig. 6.26d shows that the addition of NH3 initially produces a small amount of N2O and after some time delay also produces N2. The experiment represented by Fig. 6.26d is also consistent with a second reaction channel for N2 formation apart from the direct recombination of adsorbed nitrogen atoms. The N2 produced in this experiment is most likely due to the SCR reaction:
NOads + NH2ads −→ N2 + H2O
300 Chapter 6
This helps to explain the coproduction of H2O. The di erence in selectivity on Pt for the ammonia oxidation reaction, which produces N2 at low temperature and NO at high temperature is due to the very di erent surface composition at these conditions. At low temperature, where N2 recombination is slow, the surface is covered mainly by NHx species. Above the temperature of N2 desorption, oxygen is the dominating surfaceadsorbate species. Once NH3 decomposes then the probabilities for NO formation are high.
6.4.1 Ammonia Oxidation with Pt2+ Ion-Exchanged Zeolite Catalysts; Catalysis Through Coordination Chemistry
Ammonia oxidation can also be carried out over Pt-exchanged zeolite catalysts. In these systems, it is desirable to produce highly dispersed Pt along the exterior or inside the micropores of the acidic zeolite. This is accomplished by exchange of cations located inside the zeolite channels by positively charged Pt2+ complexes and the consecutive reduction of these complexes. The Pt2+(NH3)4 complex is typically the preferred ion of choice for carrying out the exchange with zeolite channel cations. If one wishes to prepare a highly dispersed Pt catalyst on an alumina or magnesium oxide surface, it is worth remembering that these surfaces are predominantly covered with basic OH− groups. The preferred complex for ion exchange is then PtCl4 2−. The Pt(NH3 )4 2+ ion-exchanged zeolite can be converted into an excellent low temperature oxidation catalyst for the production of
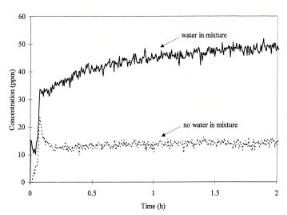
N2 from NH3. The results for conversion of ammonia in the presence and absence of H2O are shown in Fig. 6.27.[56].
Figure 6.27. The concentration of nitrogen in the oxidation of ammonia over Pt(NH3 )4 containing ZSM-5 at 473 K. Water 0%, dotted line; 1.7%, full line. The N2 O production ( 5 ppm) is una ected by addition of water[58].
In contrast to the noble metal catalysts discussed in the previous section, the presence of water in the bifunctional Pt zeolite does not suppress the activity of the reaction but actually enhances the rate of ammonia conversion and also the reaction selectivity to N2. The two primary reaction products are, once again, N2O and N2. Water enhances the selectivity for the formation of N2.
Mechanisms for Aqueous Phase Heterogeneous Catalysis and Electrocatalysis 301
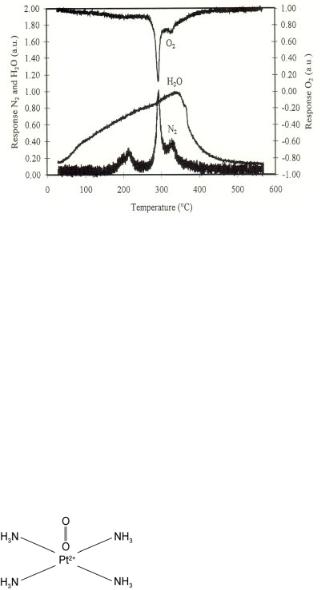
Figure 6.28. Temperature-programmed mass spectrometry of the decomposition of Pt2+(NH3 )4 ion exchanged in zeolite HZSM-5[58b]. The corresponding reaction steps are indicated. Three peaks can be distinguished in the TPD spectrum. Oxygen is consumed in only two of the N2 formation peaks. The mechanism that explains the occurrence of these three peaks is consistent with proposals made earlier on
the homogeneous oxidation of Ru–amine complexes in basic solution[59] and the reaction of NO with Ru or Os complexes[60].
Temperature-programmed desorption data reveal three di erent temperatures at which the Pt(NH3)4 2+ complex reacts with O2 (Fig. 6.28)[58].
The zeolite lattice carries a negative charge, which compensates for the positive charge of the ion-exchanged Pt complex. The zeolite lattice oxygen atoms therefore act as basic sites. At low temperature the key initial reaction step is
Pt(NH3)4 2+ + O2 −→ [Pt(NH3)3NO]+ + H+ + H2O |
(6.31) |
In the Pt–amine complex, the NO ligand formally has a charge of –1. The Pt(d8)- containing Pt2+(NH3 )4 complex was determined to be planar. Therefore, the initial reaction intermediate of O2 and Pt2+ (NH3)4 probably involves a complex similar to that showns in Fig. 6.29.
Figure 6.29. Schematic illustration of a potential intermediate that forms upon the adsorption of O2 on Pt2+(NH3 )4 (schematic).
where O2 is stabilized by the back-donation of electron density from filled molecular orbitals into dxz , dyz states. The reaction of NH3 with O2 ultimately leads to the formation of NO. This is due to the fact that a single N atom is not stabilized on a single Pt2+ center and must therefore react with other potential intermediates. After the NO formation step, five di erent reactions can occur:
302 Chapter 6
Reaction step 1:
[Pt(NH3)3(NO)]+ −→ [Pt(NH3)2] + N2 + H+ + H2O |
(6.32) |
This involves the reductive elimination of N2, which is initiated by the activation of NH3 with NO to form NH2. It is the analogue of the Fogel reaction discussed in the previous section.
Reaction step 2:
[Pt(NH3)3(NO)]+ + H2O −→ [Pt(NH3)(NO2 )]− + 2H+ |
(6.33) |
Here, Pt2+ acts as an oxidant and liberates atomic O from N2O. When followed by step 4, this intermediate will also lead to H2 production but now promoted by H2O.
Reaction step 3:
[Pt(NH3)3(NO)]+ + O2 −→ [Pt(NH3)3(NO3 )]+ |
(6.34) |
In this reaction, NO3 − formed by oxygen consumption is responsible for N2O formation (see reaction step 5).
Reaction step 4:
[Pt(NH3)3(NO2 )]− + H+ −→ Pt(NH3)2 + N2 + 2H2O |
(6.35) |
In this reaction, a proton is consumed to produce N2.
Reaction step 5:
[Pt(NH3)3(NO3)]+ + H+ + 2NH3 −→ Pt2+ (NH3)4 + N2O |
(6.36) |
Here, Pt2+(NH3)4 is regenerated by a reaction between the zeolitic protons of Pt(NH3 )2 and NH3:
Pt(NH3)2 + 2NH3 + 2H+ −→ Pt2+ (NH3)4 + H2 |
(6.37) |
The same reaction regenerates the Pt2+ (NH3)4 complex after reactions (6.32) and (6.35). In the presence of oxygen, hydrogen will be oxidized to form H2O, which drives this reaction thermodynamically. In the overall reaction scheme, N2 and N2O formation compete in the absence of water through reactions (6.32) and (6.36). In the presence of water, N2 formation is promoted because of reactions (6.33) and (6.35). There is ample evidence that the reduction of the zeolitic protons by reduced metal atoms such as Pt is actually an easy reaction step[61] . In the temperature-programmed desorption spectra (Fig. 6.28), N2O formation occurs predominantly in the peak in which excess oxygen is consumed. The other two peaks follow N2 formation. In the absence of water ,the low-temperature peak that corresponds to the reaction sequence initiated by reaction step (6.33) is suppressed.
Mechanisms for Aqueous Phase Heterogeneous Catalysis and Electrocatalysis 303
6.4.2 Electrocatalytic NH3 Oxidation
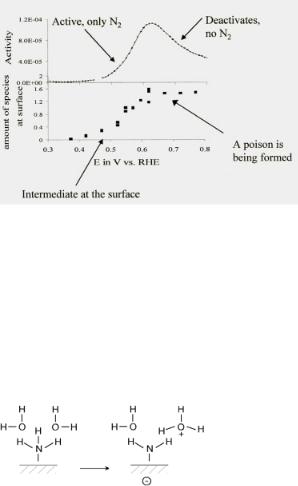
Two important factors that act to control the electrocatalytic oxidation of ammonia at room temperature are the NH3 conversion activity (current) and the build-up of surface species[61] . Figure 6.30 follows these two important features experimentally as a function of potential. The results clearly show that as the rate is increased, the surface becomes increasingly covered with surface intermediates. This occurs up to a potential of 0.6 V versus RHE, where the activity goes through a maximum. The nitrogen adatom coverage at this point approach, about 0.5 ML. Poisonous surface intermediates begin to form at potentials greater than 0.6 V and thus lower the activity.
Figure 6.30. The electrochemistry of NH3 oxidation: current against potential coverage[62].
A primary and important conclusion from this experiment is that NH3 is converted to N2 at potentials lower than where surface oxygen species are generated due to the anodic oxidation of H2O. This implies that the initial step of NH3 activation is
NH3 −→ NH2ads + Hsolvated + + e |
(6.38) |
The activation of NH3 by the reaction with a co-adsorbed oxygen atom or hydroxyl group that occurs over the metal in the gas-phase reaction is now replaced by the acceptance of protons by H2O molecules from solution (Fig. 6.31).
Figure 6.31. Protolysis reaction of ammonia.
The protons are stabilized here by solvation in solution. The key initial elementary step is heterolytic activation of NH3 to give surface NH2 − and an aqueous H3O+, a reaction that reminds us of the initial activation of NH3 adsorbed on Pt2+ by the basic
304 Chapter 6
zeolite lattice (6.32) discussed previously. For the electrochemical decomposition of H2O, theoretical results showing the analogous reaction were discussed in Section 6.1. From the slope of Tafel plots (see Addendum to this chapter), it is deduced that the rate-limiting step of the reaction is the oxidation of NH2 to a surface intermediate that, in a consecutive step, is converted to N2:
NH2ads |
→ N2Hx + (4 − x)H+ + (4−x)e (rate−determining step) |
(6.39) |
N2Hx |
→ N2 + xH+ + xe + free site |
(6.40) |
This reaction towards N2 is of course assisted by the H2O phase that can accept the protons that are released in the N2 formation step. The rate of N2 formation is 0.01 N2 mol/s/Ptatom. Adsorbed nitrogen atoms are found to lead to catalyst poisoning. They are formed in the following reaction steps:
NH2ads |
−→ NHads + H+ + e |
(6.41) |
NHads |
−→ Nads + H+ + e |
(6.42) |
The recombination of nitrogen atoms does not occur under the conditions of the electrochemical experiment. At higher potentials other reactions tend to take place. Oxygen develops at the electrode and ammonia is oxidized to N2O, NO, NO2 − and NO3 −. The selection of path (6.400 versus (6.420 correlates with the heat of adsorption of Nads. The governing mechanism over Pd, Rh, Ir and Ru metal surfaces is similar to that which is seen on Pt. The steps outlined in (6.40) do not appear to occur over Pd, Rh or Ru. Reaction (6.42) is much faster than reaction (6.40) and therefore tends to dominate over these metals. These catalysts appear to be selective catalysts for the oxidation of NH3 to the oxidized products.
Other metals such as gold, silver or copper are not active for either reaction (6.40) or (6.42). Instead, the metal tends to undergo oxidative dissolution. It is of interest to compare the potentials at which the nitrogen adatoms poison the surface versus their heat of adsorption.
The computed sequence of decreasing Nads binding energies is as follows:
Ru > Ir > Rh > Pd > Pt > Cu > Au > Ag
The electrochemical experiments indicate the following sequence measured according to the potential at which the metal poisons:
Ru > Rh > Pd > Ir > Pt >> Cu, Au, Ag
The two trends are quite similar, with Ir being the exception.
Here we have shown that the presence of water and electrochemical potential can significantly alter both the activity and the selectivity of the NHx reaction paths. Care must be taken in comparing the results obtained between the vapor phase and those under electrochemical conditions in the electrochemical experiments. Second, explicit evidence has been found that NHx species are involved in the N2 formation reaction.
Mechanisms for Aqueous Phase Heterogeneous Catalysis and Electrocatalysis 305
6.5 Electrochemical NO Reduction
As was noted in Chapter 3, at the vapor metal surface, interface the dissociative adsorption of NO is strongly site dependent. The activation barriers for NO dissociation are significantly reduced at step edges. Such a dependence does not appear to play a role in electrochemical reduction experiments on NO. Product selectivity is sensitive to the presence or absence of NO in solution. On polycrystalline Pt, the reduction of adsorbed
NO in the presence of NO in the solution yields N2O as the only product at potentials between 0.4 and 0.8 V vs RHE[63].
The mechanism for this reaction is not of the Langmuir–Hinshelwood form, but rather involves the reaction between a surface-bonded NO molecule with an NO molecule in the solution phase along with a simultaneous electron transfer. The Tafel slope in acidic solution is (120 mV)−1, which implies that the first electron-transfer step is rate-determining (see Addendum to this chapter). The reaction is first order in H+ and shows no apparent
isotope e ect. The following mechanism has been proposed: |
|
|
NOaq −→ NOads |
(fast) |
(6.43) |
NOads + NOaq + H+ + e −→ HN2O |
(rds) |
(6.44) |
HN2O2 + H+ + e −→ N2Oaq + H2O |
(fast) |
(6.45) |
The rate-determining step (rds) consists of a pre-equilibrium involving the protonation followed by a rate-determining electron transfer step.
Stripping voltammetry of adsorbed NO in acidic solutions exhibits three peaks at 0.15, 0.23 and 0.7 V vs RHE. The only product formed is NH4 +. The third peak vanished when the SO4 2− in solution is replaced by ClO4 −. This suggests that two types of surface-bound NO exist.
The voltammetric results on stepped and non-stepped surfaces appear to be very similar[64]. The Tafel slopes of the first two peaks in the adsorbate solution is ca (40 mV)−1 which is typical for the electrochemical equilibrium followed by a rate-determining potential-independent chemical step. The following reaction scheme is proposed:
NOads + H+ + e |
−→ HNOads |
(equilibrium) |
(6.46) |
HNOads + H+ + e |
−→ H2HNOads |
(rds) |
(6.47) |
H2NOads + 4H+ + 3e |
−→ NH4 + + H2O |
(fast) |
(6.48) |
HNO is suggested as the hydrogenated intermediate rather than NOH because gas-phase HNO is nearly 100 kJ/mol more stable than gas-phase NOH. H2NO is chosen over HNOH as the second hydrogenated intermediate, since calculations show that gas phase H2NO is over 500 kJ/mol more stable than HNOH. No direct activation by the surface is involved.
All metals show a high selectivity to N2O at higher potentials and a high selectivity to NH3 at low potentials. N2 is predominantly formed at intermediate potentials. The formation of N2 produced at potentials between the formation of N2O and NH3 most likely takes place by the reduction of previously formed N2O. The activation of NO in the electrochemical system is very di erent from that which takes place over the metal in the gas phase, in that there may not be a beneficial e ect of the presence of steps on the kinetics.