

Molecular Heterogeneous Catalysis, Wiley (2006), 352729662X
.pdf266Chapter 5
98.M.A. Johnson, E.V. Stefanovich, T.N. Truong, J. Phys. Chem. B, 101, 3196 (1997)
99.R. Orlando, F. Cora, R. Millini, G. Perego, R. Dovesi, J. Chem. Phys. 105, 351 (1999)
100.K.J. Borve, L.G. Pettersson, J. Phys. Chem. 95, 3214 (1991)
101.M.L. Anchell, K. Morokuma, A.C. Hess, J. Chem. Phys. 99, 6004 (1993)
102.J.M. DeBoy,R.R. Hicks, J. Chem. Soc. Chem. Commun. 982 (1988)
103.M.S. Palmer, M. Neurock, M.M. Olken, J. Phys. Chem. B, 106, 6543 (2002)
104.K. Otsuka, A.A. Said, K. Jinno, T. Komatsu, Chem. Lett. 77 (1987)
105.K. Otsuka, Y. Murakami, Y. Wada, A.A. Said, A. Morikawa, J. Catal. 121, 122 (1990)
106.C. Lin, K.D. Campbell, J.X. Wang, J.H. Lunsford, J. Chem. Soc. Chem. Commun. 90, 534 (1986)
107.J.X. Wang, J.H. Lunsford, J. Phys. Chem. B, 90, 3890 (1986)
108.M Banares, Catal. Today, 51, 319 (1988)
109.E. M. Kennedy, N.W. Cant, Appl. Catal. 75, 321 (1991)
110.V.T. Amorebieta, A.J. Colussi, J. Am. Chem. Soc. 118, 10236 (1996)
111.A. Shamsi, Ind. Eng. Chem. Res. 32, 1877 (1993)
112.R. Burch, G.D. Squire, S.C. Tsang, Appl. Catal. 43, 105 (1988)
113.J.V. Lauritsen, M.V. Bollinger, E. Lægsgaard, K.W. Jacobsen, J.K. Nørskov, B.S. Clausen, H. Topsøe, F. Besenbacher, J. Catal. 221, 510 (2004)
114.J. Raybaud, J. Hafner, G. Kresse, H. Toulhoat, J. Phys. Condens Matter, 9, 11107 (1997)
115.J. Raybaud, J. Hafner, G. Kresse, , S. Kasztelan,H. Toulhoat, J. Catal. 190, 128 (2000)
116.G. Wul , Z. Kristallographic, 34, 449 (1901);
W.K. Burton, N. Cabrera, F.C. Frank, Philos. Trans. Roy. Soc. A, 243, 299 (1951)
117.(a) L.S. Byskov, J.K. Nørskov, B.S. Clausen, H. Topsøe, J. Catal. 187, 109 (1999);
(b) P. Raybaud, J. Hafner, G. Kresse, S. Kasztelan, H. Toulhoat, J. Catal. 189,
129 (2000)
118.J.V. Lauritsen, M. Nyberg, J.K. Nørskov, B.S. Clausen, H. Topsøe, E. Lægsgaard, F. Besenbacher, J. Catal. 224, 94 (2004).
119.A. Travert, H. Nakamura, R.A. van Santen, S. Cristol, J.-F Paul, E. Payen,
J. Am. Chem. Soc. 124, 7084 (2002)
120.E.J.M. Hensen, G.M.H.J. Lardinois, V.H.J. de Beer, J.A.R. van Veen, R.A. van Santen, J. Catal. 187, 95 (1999)
121.E.J.M. Hensen, H.J.A, Brans, G.M.H.J. Lardinois, V.H.J. de Beer, J.A.R. van Veen, R.A. van Santen, J. Catal. 192, 98 (2000)
122.H. Kn¨ozinger, S. Huber, Faraday Trans. 94, 2047 (1998)
CHAPTER 6
Mechanisms for Aqueous Phase Heterogeneous Catalysis and Electrocatalysis; A Comparison with Heterogeneous Catalytic Reactions
6.1 General Introduction
A wide range of di erent heterogeneous catalytic reactions which are carried out in liquid or aqueous media show marked changes in activity and/or selectivity over the same reactions carried out in the vapor phase. The rate can be reduced owing to reactant availability at the catalyst surface as a result of external mass transfer or solubility limitations. These are classical, well-characterized extrinsic kinetic limitations. The solution phase can also act to alter the intrinsic chemical kinetics associated with the elementary adsorption, surface reaction, surface di usion and desorption processes. It is well established in physical organic chemistry that the solution phase can influence a reaction by stabilizing or destabilizing its transition and products states over those of the reactant state. For example, both aqueous and polar solvents tend to stabilize reactions that have transition states which are more polar than their corresponding reactant state. The aqueous or polar medium stabilizes a charge transfer process and the corresponding transition state that forms. Similarly, the presence of an aqueous phase or polar medium tends to enhance heterolytic bond activation reactions that result in electron transfer and/or proton transfer processes. These reactions are at the heart of both electrocatalysis and enzyme catalysis. In addition to their influence on charge stabilization or destabilization, the aqueous medium or protic solvent can also directly participate in the catalytic action via proton and electron transfer processes.
A fundamental understanding of the atomic and electronic processes that govern reactions in solution has eluded the heterogeneous catalysis community to a large extent owing to the complexity of the aqueous medium and its interaction with heterogeneous substrates. Previous experimental e orts aimed at characterizing the interface between the metal and an aqueous solution, for example, have been met with very limited success owing to the di culty in resolving molecular scale information. The critical spectroscopic bands that might be used to follow specific reaction modes are typically masked by the spectral features that arise from the bulk solution.
The past decade, however, has witnessed the development of a host of spectroscopic methods that can begin to elucidate molecular structure at the aqueous/metal interface. Surface Enhanced Raman Spectroscopy (SERS), Surface Enhanced Infrared Spectroscopy (SEIRS), and Sum-Frequency-Generation (SFG) can begin to separate out the surfaceadsorbate characteristics that arise fromfrom those from the bulk solution and, thus provide the vibrational properties of molecules adsorbed on a metal substrate to be followed along the course of a catalytic reaction. In addition, the exponential growth of computational resources along with novel algorithm development has made it possible to begin to simulate the surface structure and the elementary processes that occur at the metal/solution interface.
In this chapter we extend our treatment of mechanisms for metal-catalyzed reactions in the vapor phase to heterogeneous catalytic reactions carried out in aqueous media and electrocatalytic reactions. More specifically, we discuss what is known about the water/metal interface, its reactivity, and the influence of the aqueous phase on elementary surface processes including adsorption, reaction, di usion and desorption and solutionphase kinetic processes. We advance these ideas into the discussion of the mechanisms
Molecular Heterogeneous Catalysis. Rutger Anthony van Santen and Matthew Neurock Copyright © 2006 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
ISBN: 3-527-29662-X
268 Chapter 6
that govern four di erent example reactions, namely the synthesis of vinyl acetate, the low-temperature oxidation of ammonia, NO reduction, and CO oxidation. We will draw ties between organometallic coordination complexes and their reactivity, gas-phase heterogeneous catalysis, heterogeneous catalysis in solution and electrocatalysis.
6.2 The Chemistry of Water on Transition Metal Surfaces
The structure and chemistry at the water/metal interface is critical in dictating the properties and ultimately controlling the catalytic performance of aqueous phase heterogeneous, electro-, homogeneous, and bio-catalytic systems. In addition, they have great relevance to understanding corrosion as well as a host of other materials issues. A number of outstanding reviews exist which describe the structure and reactivity of water over metal and metal oxide substrates[1,2] . Rather than repeat these analyses, we try to summarize some of the important factors and describe the systematic changes that occur in the structure, adsorption and reactivity of water on metal substrates as we move from the adsorption of water in the vapor phase to the solution phase and then on to the influence of applied potentials.
6.2.1 Reactions in Solution
The presence of solution can dramatically alter the resulting chemistry at the solutionmetal interface. This is clearly present even in the neat liquid phase dissociation processes alone. For example, the dissociation of acetic acid proceeds in the vapor phase at higher temperatures via a homolytic process that leads to the formation of CH3CO2• and H• free radical intermediates. This homolytic activation of the O–H bond in acetic acid costs 440 kJ/mol[3a]. The heterolytic activation of acetic acid to form CH3CO2– and H+ intermediates, however, is significantly more endothermic, costing +1532 kJ/mol.
This reaction is simply the reverse of the proton a nity for acetate. The dramatic increase in the endothermicity for the heterolytic reaction is due to the fact that the charged products which form are unstable alone in the vapor phase. The neighboring water molecules begin to hydrogen bond with acetic acid and stabilize the charged complexes. The results in Fig. 6.1 indicate that there is a precipitous drop in the endothermicity of the dissociation reaction that occurs by adding just 1, 2, or 3 water molecules. The water molecules e ectively stabilize both the proton and the acetate anion (CH3COO−) that form. As we get to 12 water molecules or more, the dissociation actually becomes thermoneutral. The stabilization of the charged products is much stronger than the energy required for the heterolytic activation, hence the dissociation energy becomes thermoneutral. The dissociation energy changes from +1532 kJ/mol in the vapor phase to 0 kJ/mol in solution. This agrees quite well with experimental results, which indicate that the proton a nity is 1540 kJ/mol whereas the dissociation of acetic acid in solution is 1.6 kJ/mol.
6.2.2 The Adsorption of Water on Metal Surfaces
The adsorption of water on most metal surfaces is typically rather weak and controlled by a balance between the strength of the metal–water bond and the water–water[1,2,4,5] interactions. Molecular water adsorbs on metal and metal oxide substrates through the donation and back-donation of electrons between the frontier molecular orbitals of water and the states of the metal near the Fermi level.
Mechanisms for Aqueous Phase Heterogeneous Catalysis and Electrocatalysis 269
Figure 6.1. The e ect of water on the energies for the heterolytic dissociation of acetic acid. (a) Gas phase, (b) reaction with one water molecule, (c) reaction with three water molecules, (d) reaction in bulk water.
Figure 6.2. Molecular orbitals and their corresponding energies for water.
On metal oxides weak hydrogen bond interactions can also evolve. The molecular orbital energy diagram for the gas-phase water molecule alone is shown in Fig. 6.2.
The frontier orbitals are the 1b1, 3a1 and the 1b2 states. We recognize these orbitals as two px- and py -type oxygen atomic orbitals interacting with symmetric and antisymmetric hydrogen atomic orbital combinations (1b2 and 3a1, respectively). The 1b1 molecular orbital is the non-bonded 2pz atomic orbital on oxygen. All three of these states are
270 Chapter 6
typically seen in valence band photoemission spectroscopy. Which of these states controls adsorption is di cult to discern from the shifts in photoemission spectroscopy alone [2] .
Michaelides and co-workers[4,5] have shown theoretically that for monolayers and bilayers of water on Ru, the predominant overlap occurs between the 2px type 3a1 orbital of water with a dz2 state on Ru. In addition there is overlap between the lone–pair 1b1 orbital on water and the Ru dz2 state. The overlap and mixing of the metal surface state with the 1b1 orbital is significantly stronger. An electronic analysis shows that there is a charge depletion from the 1b1 and Ru dz2 states with a charge accumulation on the
lower lying dxz and dyz states. There is also a small charge increase between the O and the Ru[4].
The adsorption of water at low coverage on most metal surfaces is fairly weak, whereby water prefers to sit atop a metal atom so as to avoid Pauli repulsive interactions between the lone pair of electrons on water and the filled states of the metal. The bond lengths between the metal and the oxygen are within the range 2.1–2.3 ˚A[3b,5]. Valence bond theory indicates that water should tilt significantly with respect to the surface. The degree of tilt with respect to the surface normal varies with the metal and the nature of the surface. At low coverages, water can adsorb as isolated species; form 2D or 3D clusters; assemble into well-ordered surface structures commensurate with the registry of metal lattice spacing; or dissociate into OH and H. The structures that form depend upon the balance between the metal–water bond strength and water–water bond strength for the specific system of interest in addition the system conditions, i.e. temperature and pressure. For systems where the metal-water interactions are weaker than the water–water interactions, water prefers to form clusters. For systems where the metal–water interactions are stronger than the water–water interactions, water will tend to form commensurate surface structures. For systems where the metal-water interactions are much stronger than the water–water interactions, water can begin to dissociate[2] .
The adsorption of water onto the metal in the vapor phase is typically quite weak with adsorption energies on the order of 30–50 kJ/mol, with some notable exceptions[3b,5] . DFT slab calculations predict the adsorption of a single water molecule to be 50 kJ/mol on Ag(111), 30 kJ/mol on Pd(111), 30 kJ/mol on Pt(111), 37 kJ/mol on Rh(111), and 100 kJ/mol on Cu(110). Noteworthy is the increased hydrophilicity of the Group IB noble metals compared with that of the group VIII metals.
As the coverage is increased, water can form monolayers, bilayers or 3D water[2] clusters. In addition, water can form either ice-like crystalline surface structures or amorphous liquid water. This, once again, is highly dependent upon the balance between metal–water and water–water bond strengths and also the system conditions. Vassilev et al.[6], for example, found that as the coverage of water was increased from 1/3 to 2/3 ML, the e ective adsorption energy per water molecule increased from 37 to 56 kJ/mol. At 2/3 ML coverage, the water layer is actually a bilayer which takes on considerable hydrogen-bonding interactions between neighboring water molecules. When the binding energy of the entire bilayer is calculated, it is found that the interaction energy with the metal surface is essentially vanishing. As mentioned above, a very similar conclusion follows from the work of Desai and Neurock[3b] .
The adsorption of water on most close-packed metal surfaces leads to the formation of the well-known bilayer structure. In this system, water prefers to adsorb on the metal substrate in a hexagonal ring structure which matches the registry of the metal substrate, as shown in Fig. 6.3. Three water molecules within the ring are bound to the substrate through their oxygen ends. The two hydrogen atoms are directed towards the oxygen

Mechanisms for Aqueous Phase Heterogeneous Catalysis and Electrocatalysis 271
end of two neighboring water molecules, thus forming two hydrogen bonds to the solution phase. Each of the three water molecules bound to metal surface within the hexagonal ring brings in one more water molecule due to hydrogen bonding, thus resulting in six water molecules that make up the hexagonal ring structure. These secondary waters are further away from the surface where they form hydrogen bonds with those water molecules that are directly bound to the substrate.
Figure 6.3. The general bilayer structure of water adsorbed on a close-packed transition-metal surface.
(a) H-up structure, (b) H-down structure, (c) transition state for bilayer dissociation, and (d) partially dissociated bilyaer structure[4].
This leads to the formation of two distinct layers of water molecules[2,5] . The first layer is directly bound to the surface via metal oxygen bonds whereas the second layer is indirectly bound to the substrate via the formation of hydrogen bonds. Two di erent conformations of the bilayer are possible. In the first conformation, one of the hydrogen atoms from the water molecules in the second layer is pointed up (H-up) into the vacuum. In the second, the hydrogen atom from the water molecule in the second layer is directed down toward the metal substrate (H-down).
Water which is bound to most transition-metal surfaces tends to form this bilayer structure on close-packed surfaces. Experimental results for water on Ru(0001), however, were found to be characteristically di erent than those on other transition-metal surfaces. Held and Menzel[7] identified the oxygen atoms from water molecules in two di erent
layers and speculated on the formation of a “bilayer” structure for water adsorbed on
√ √
Ru(0001) in a ( 3 x 3)R30◦ state separated by only 0.1 ˚A[7]. They speculated that this was the result of the formation of a compressed water bilayer. Feibelman[8] later showed via theoretical calculations that the only water structures that fit the short 0.1 ˚Aseparation distance between oxygen atoms of the bilayers are those that involve partially dissociated water along with neighboring water molecules. Feibelman[8] and Michaelides et al.[4] showed that the barrier for water dissociation was reduced from 0.8 eV to 0.5 eV of moving from isolated water molecules to water molecules contained within the bilayer structure. The partially dissociated states also had the lowest energies. Water can also dissociate over various other metal surfaces, especially at higher temperatures.
The dissociation of an isolated water molecule occurs via the insertion of metal atom into one of its O–H bonds. As the O–H bond is initially activated, the energy level of the unoccupied σ antibonding O–H orbital lowers, thus allowing for electron transfer from the metal into this state. This subsequently facilitates the activation of water. Dissociation of an isolated water molecules adsorbed from the gas phase at the metal/vapor interface leads to the formation of a surface hydroxyl as well as a surface hydride. Desai and Neurock[3b] showed that the activation of water over Pt(111) would be highly endothermic (+90

272 Chapter 6
kJ/mol) and require overcoming a barrier of +140 kJ/mol. The reaction path is shown in Fig. 6.4 (see also Chapter 3. pages 133, 134).
Figure 6.4. The homolytic activation of water over Pt(111). On the left: water adsorbed reactant state.
In the center the transition state for O–H activation. On the right the surface hydroxyl and hydride products[3b].
Under UHV conditions, water would preferentially desorb rather than react. The barrier for the activation of water over Ru, however, is considerably lower at +90 kJ/mol. The reaction is now slightly exothermic. The barrier, however, is still too high to overcome under UHV conditions. The barrier however is reduced significantly as the density of water increases from the monomer structure to the bilayer and then on to multilayer adsorption, as will be discussed.
The dissociation of water was examined over a range of di erent close-packed transitionmetal surface structures in order to establish periodic trends. The results shown in Fig. 6.5 indicate that it becomes easier to activate water over metals that lie to the left of the periodic table which have more vacancies in the d-band.
Figure 6.5. Periodic trends in the reaction energies for the homolytic surface dissociation of water at the vapor/metal interface. Energies are in kJ/mol.
In addition, water is also more readily activated over metals that lie higher up in the periodic table. The relative ordering of the metals, examined in Fig. 6.5, that most readily activate water is:
Ni > Ru > Rh > Cu
The activation of water at the aqueous metal interface can be characteristically di erent from that at the vapor metal interface owing to changes in the dielectric constant of the medium, increased hydrogen bonding network and potential changes to the metal substrate. Density functional theoretical results for multilayers of water adsorbed on Pt(111) indicate that the water molecules near the surface take on a bilayer-like structure whereas
Mechanisms for Aqueous Phase Heterogeneous Catalysis and Electrocatalysis 273
water molecules in solution either form an ordered ice-like structure if they are optimized at 0 K, or form a more amorphous random structure as the result of ab initio MD simulations at higher temperatures. The homolytic activation of water to form adsorbed hydrogen and a surface hydroxyl intermediate is stabilized owing to the presence of aqueous water but the change is only 10 kJ/mol. This is likely due to the fact that the transition state for the homolytic activation is not very polar. A second, more likely, path would involve the heterolytic activation of water to form a proton (H+ ) which is stabilized by the aqueous medium and a negative hydroxide ion on the metal substrate. The extra electron can readily transfer to the metal. The proton migrates away from the surface via proton transfer through the network of water molecules at the metal surface. Water directly participates in the mechanism by providing a conduit for proton transfer. This lowers the barrier by over 40 kJ/mol. The reactant, transition and product states for this reaction over Pt(111) are very similar to those over the Pt66.6%Ru33.3% surface which, is shown in Fig. 6.6[3b].
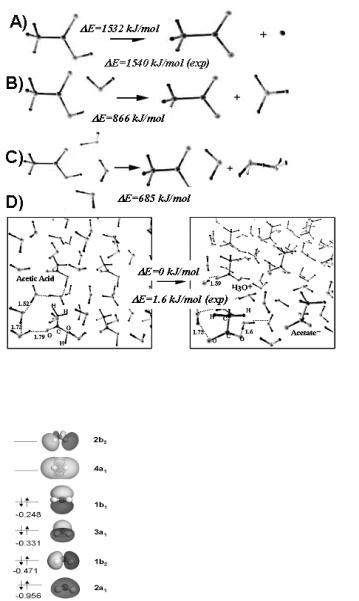
Figure 6.6. The heterolytic activation of water over Pt66.6% Ru33.3% alloy in the presence of aqueous water. (a) Water adsorbed reactant state, (b) the transition state for O–H activation, and (c) the surface hydroxyl and aqueous hydronium ion product states.[3b]
The addition of Ru to the Pt surface can influence both the vapor phase and the aqueous phase reaction energies.[3b] The substitution of just 1 out of every 3 Pt atoms in Pt surface layer leads to a significantly lower activation barrier. The results in the vapor phase indicate that the barrier falls by nearly 45 kJ/mol to 108 kJ/mol from the Pt(111) surface. This is due to the fact that water preferentially adsorbs and activates at the Ru sites. The activation barrier for water over the Pt66.6%Ru33.3% surface is 106 kJ/mol, which is only 11 kJ/mol higher than that on the Ru(0001) surface. This is due to the fact that the reaction is local, involving a single metal atom insertion. The surrounding Pt atoms have a weak electronic e ect on the chemistry at the Ru site. The activation of water over the Pt66.6%Ru33.3% alloy in an aqueous medium, however, is quite di erent. The reaction proceeds heterolytically, thus forming a solvated proton, a surface hydroxyl intermediate and an electron that is delocalized over the metal. The transition state for the heterolytic reaction is much more polar and therefore can be stabilized by the presence on an aqueous medium. The barrier for this reaction over the alloy surface in solution is only 26 kJ/mol. This is considerably lower than the energy for the vapor phase reactions and those reactions carried out over Pt and Ru substrates in the aqueous phase. There appears to be strongly enhanced synergy between Pt, Ru and the aqueous medium. The reactant, transition, and product states for this reaction are shown in Fig. 6.7.
274 Chapter 6
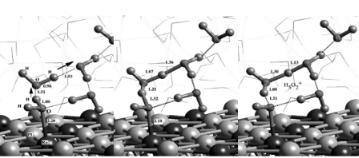
Figure 6.7. The adsorption of atomic hydrogen on (A) Pt(111) in aqueous solution leads to proton formation whereas the adsorbed atomic hydrogen on (B) Ru(0001) remains as adsorbed hydrogen in the presence of an aqueous solution. [3b]
Figure 6.8. The overall Born–Haber thermodynamic cycle for the free energy required to create hydronium ions from adsorbed hydrogen in the presence of water[3b,10].
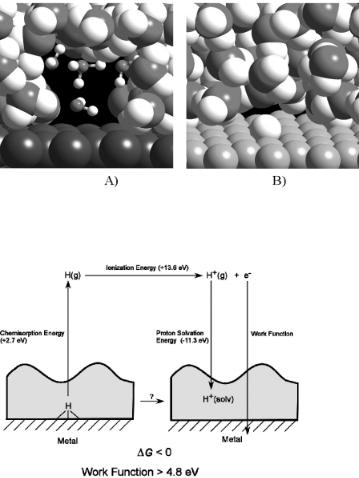
A key step in the aqueous phase activation of water involves the generation of protons. Ab initio calculations showed that atomic hydrogen adsorbed on Pt(111) would transfer its electron into the metal and then migrate into solution as a proton. Wagner and Moylan[9] and later Kizhakevariam and Stuve[10] showed experimentally that adsorbed hydrogen in the presence of solution on Pt(111) forms hydronium ions. Similarly, Desai and Neurock[3b] used theory to show that adsorbed hydrogen on Pt(111) can readily transfer its electron to the metal and thus generate protons. The net results, however, would ulrimately require that proton and the electron recombine. The results from this reaction are shown in Fig. 6.7A. These same calculations performed on the Ru(0001) surface indicate that the hydrogen remains bound to Ru as a hydride (Fig. 6.7B). These di erences can be explained by constructing a simple Born–Haber cycle for the process such as that shown in Fig. 6.8:
H(a) + H2O(aq) + M −→ H3O+ (aq) + M + 1 |
(6.1) |
Mechanisms for Aqueous Phase Heterogeneous Catalysis and Electrocatalysis 275
The desorption of hydrogen and its dissolution into the aqueous phase require the following steps: (1) desorption of H• into gas phase, (2) the ionization of H+ , (3) the solvation of the proton into water, and 4) the capture of the resulting electron by the metal (i.e. electron a nity). The only steps which change on moving to di erent metals are the desorption of hydrogen and the work function of the metal. Steps 2 and 3 are identical regardless of the metal. While the desorption of hydrogen changes with the metal substrate used, the change is small compared with the changes in the work function of the metal. Kizhakevariam and Stuve[10] estimated that the free energy for proton transfer would be greater than or zero for metals with work functions greater than 4.88 eV. Theoretical results from Desai and Neurock[3b] showed that Pt and Pd in the presence of adsorbed water had work functions that were greater than 4.88 eV. The overall energy for proton formation was calculated to be –57 and –14 kJ/mol exothermic over Pt(111) and Pd(111) substrates, respectively. The work function for Ru(0001) was calculated to be below 4.88 eV and thus the reaction to form the hydronium ion was found to be endothermic by +40 kJ/mol. This suggests that while Ru may activate water it will have a stronger tendency to hold onto the hydrogen atoms as a hydride.
Interestingly, the Pt66.6%Ru33.3% surface alloy has a work function that is 4.92 eV, which would suggest that this metal could result in the formation of protons. The calculations confirm that hydrogen atoms desorb as protons on this surface. The key here is that the addition of Pt to the Ru lattice helps to aid in the heterolytic activation of water since Pt increases the work function of the metal, thus enhancing its ability to accept electrons. The Pt, Ru and aqueous solution form a unique active nanoscale environment whereby Ru is necessary to adsorb and activate water, Pt increases the work function of the metal which enhances electron transfer, and finally the aqueous solution phase promotes proton transfer into solution.
It is clear that the activation of water at the aqueous phase/metal interface is quite di erent than the activation of water at the vapor phase/metal interface owing to charge stabilization and electron transfer. The ideas presented on PtRu can be extended to the activation of water in the aqueous phase over various other metals. The results in Fig. 6.9 were calculated by using a Born–Haber cycle similar to that shown for hydrogen dissolution described earlier[11]. In this analysis, the ionization energy of H•, the solvation energy for H+ , the work function of the metal and the change in hydrogen-bonding contributions are used to correct the energies for the vapor-phase activation of water to account for the e ects of solution. These thermodynamic estimates of the overall reaction energies for the dissociation of water at the aqueous/metal interface are shown in Fig. 6.9 for various close-packed transition-metal surfaces. The best metals are those which demonstrate favorable energies for the vapor-phase activation of water and, in addition, have work functions that are high enough to promote the heterolytic activation of water. The best metals show the following trend with respect to their overall favorability to activate water:
Ni > Rh > Ru > Cu
The higher work function for Rh over Ru here changes the relative ordering of the metals in terms of favorability from that found for vapor-phase activation. Actual ab initio calculations which explicitly examine the reactants and products at the metal/solution interface yield overall energies for the dissociation of water on Cu and Pt in an aqueous medium of 30 and 90 kJ/mol, respectively. These values are consistent with the estimates