

Molecular Heterogeneous Catalysis, Wiley (2006), 352729662X
.pdf236 Chapter 5
Figure 5.15. The optimized BCC structure of the H3PW12 O40 .6H2O hexahydrate form. In this structure, all Od atoms are bound to an H5 O2 + species; however, many of these species have been omitted
for simplification. (A) The view of one face of the BCC structure. (B) The relationship between KUs on an edge of the cube and the nearest-neighbor body-centered Keggin unit[25].
The high Brønsted acidity found for heteropolyacids is further confirmed by ammonia adsorption studies on isolated anhydrous HPW structures[27] . The adsorption of ammonia on Moand W-based heterpolyacids were compared. The calculations of ammonia bound to the trimer HPA complex reveals chemisorption energies of –103 and –141 kJ/mol for the HPMo and HPW structures, respectively, comparable to values found for zeolites with a low concentration of alumina. The results compare very favorably with the values from microcalorimetric NH3 adsorption experiments. Ammonia adsorbs in a bidentate configuration, thus leading to a proton transfer and the formation of the ammonium ion upon adsorption. Ammonium formation tends to occur when ammonia adsorbs at the higher coordination sites. The energy cost required for charge separation must be compensated for by the stabilizing electrostatic interactions [27]. The weaker acidity for
Catalysis by Oxides and Sulfides 237
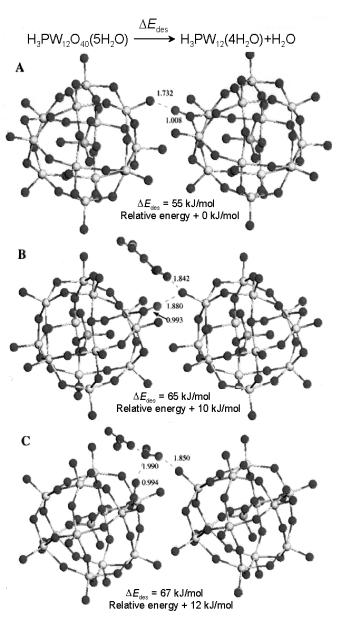
Figure 5.16. Structures resulting from the removal of two of the six water molecules of hydration from H3 PW12 O40 .6H2O. The remaining anhydrous H+ atom locations are at (A) bridging an Od atom and an Oc atom of the nearest-neighbor body-centered KU, and (B) bridging an Ob atom and an oxygen atom of a water molecule of hydration bound to the nearest-neighbor body-centered KU. Relative energies are
given with respect to the optimal configuration (A). All distances are given in ˚a. There are two remaining H5 O2 + species per KU and only those in close proximity to the anhydrous proton are shown[25].

238 Chapter 5
the HPMo compared with that of the HPW is consistent with the higher ionization potentials for sixth row transition metals compared with third row transition metals and relates to partial screening of nuclear charge by the atomic f-electrons.
The acidity of the PW12O40 3− is such that it can stabilize the adsorption of an alkene. Janik et al.[28] have shown that the barriers for the protonation of ethylene, propene, 2-butene and isobutene are 69, 50.5, 51 and 14 kJ/mol, respectively. The results indicate that the formation of the carbenium ion from the π adsorbed state is favored on the Keggin structure over the similar corresponding state in the chabazite (see Chapter 4). For example, the barrier for the protonation of propene in chabazite was calculated to be +56 kJ/mol in comparison with the value of 50.5 kJ/mol calculated on the phosphotungstic (HPW) Keggin structure. The barriers, however, for the reaction of a surface alkoxy to the carbenium ion state are all higher on phosphotungstic acid than on chabazite.
The barriers for formation of ethylene, propene, 2-butene and isobutene from the alkoxy HPW states were calculated to be 114, 93.7, 87.8 and 64 kJ/mol, respectively, as compared with the value of 83 kJ/mol for propene over chabazite. Cleavage of the alkoxide bond initiates consecutive reaction steps necessary for many hydrocarbon conversion reactions.
The catalytic di erences between protonic zeolites and the HPW structures are likely due to the greater stability of the alkoxide reactant state on the HPW structure than that on chabazite.
Table 5.4. Calculated proton a nity values for progressive addition of three protons to each of the oxygen types of PW12 O40 3−[26]
Proton a nity (kJ/mol)
Site |
1st proton |
2nd protona |
3rd protonb |
Ob |
1579 |
1349 |
1077 |
Oc |
1591 |
1340 |
1079 |
Od |
1564 |
1321 |
1071 |
|
|
|
|
a The calculations for finding the position of the second proton were performed with the 1st proton on its preferred Oc site.
b The 3rd proton calculations were performed with both the 1st and 2nd on their preferred Oc and Ob sites.
5.6 Oxidation Catalysis
5.6.1 Introduction
Metal oxides are quite diverse and posses a wide range of di erent properties which make them active materials for the conversion and selective oxidation of a wide range of di erent reagents. They can be used as acid, base, reducible, non-reducible and bifunctional catalytsts to carry out specific reactions. The mechanisms that govern the reactions over these materials, however, can be quite di erent. Here we will predominantly discuss hydrocarbon oxidation. The selective oxidation of hydrocarbons tends to proceed via a sequence of elementary steps which include: the activation of a C–H bond, oxygen insertion
into activated hydrocarbon intermediate, subsequent C–H activation steps, desorption of products, and the regeneration of the active site[29]. The initial C–H activation step
is thought to be the rate-determining step for a range of di erent oxidation processes.
Catalysis by Oxides and Sulfides 239
Subsequent oxidation and C–H activation steps, however, tend to control the selectivity and the slate of final products that form. C–H activation can proceed through either homolytic or heterolytic processes depending upon the catalyst that is used and the nature of the active site. Homolytic C–H activation processes usually lead to the production of free radical intermediates whereas heterolytic activation leads to the formation of charged complexes. The heterolytic activation of a C–H bond can occur through the formation of a carbanion intermediate which is bound to a surface metal cation and a proton which binds to a nearby surface oxygen atom, thus forming a surface hydroxyl intermediate. Alternatively, the heterolytic activation of the C–H bond can lead to the formation of a metal hydride (M–H) and a surface alkoxide intermediate.
The overall selectivity for oxidation depends not only on the composition of the catalytic material, but also on the type of active oxygen species that are present at the surface. Oxygen on the surface can exist in various di erent forms ranging from adsorbed O2 to fully oxidized atomic oxygen (O2−) as is shown in the Eq. (5.1) below [29−32].
O2 < O2− < O− < O2− |
(5.1) |
The first three of these oxygens (O2 , O2 −, O−) are typically electrophilic. They tend to activate C–H bonds and lead to total oxidation of the reactants. O2−, however, is more nucleophilic and can insert into the carbon–carbon bonds of the reactant molecules, thus favoring more selective oxidation paths.
The activation path along which one proceeds is strongly dependent on the nature of the active catalyst and the support that is used. As mentioned already, selective oxidation can occur over both reducible and non-reducible oxides. Oxidation over reducible oxides undergoes direct O2 activation at a metal center, thus leading to changes in the oxidation state of the metal. The metal is subsequently reduced upon reaction with the hydrocarbon. The active catalyst then cycles between reduced and oxidized states. Kulkarni and Wachs[33], for example, have shown that redox activity over di erent metal oxides can vary by over six orders of magnitude. Catalysis over non-reducible oxides, however, does not involve a change in the oxidation state of the metal. Instead, it requires the presence of promoters, defect sites, or sites on the oxide that do not require changes in the redox character of the cations[34] to help activate hydrocarbon reagents. Regardless of which path is ultimately taken, there are a number of properties for the di erent oxide materials that can ultimately be tuned to influence the actual catalytic behavior. The factors that typically control the properties of these catalytic systems, ultimately dictating the specific chemistry that occurs, depend upon the structure, composition, and electronic properties of the metal oxide surface. We describe here the most direct e ects on catalysis which arise from changes in the structural or electronic features of the active site directly and the e ects of changing the overall environment of the system. This includes:
1) Degree of coordination of the active site.
In many instances the metal ions can be thought of as individual or isolated molecular centers. As such, a number of the fundamental rules established from organometallic chemistry apply[35]. The relative degree of coordination, or lack thereof, is important in controlling the metal–adsorbate bond strength. This can govern the relative propensity for either C–H activation or subsequent oxygen insertion. Sites which are more coordinatively unsaturated will, in general, bind adsorbates more strongly and thus aid in the activation of adsorbate bonds. Analogously, sites which are more coordinatively saturated will bind adsorbates more weakly and tend to favor bondmaking processes.
240 Chapter 5
2) Band gap of the oxide.
The band gap of the oxide is important in that it is a measure of redox capabilities of the actual oxide. It describes the electronic propertiesy of catalytic oxide material that “align” the lowest energy states in their conduction band and the highest energy states of the valence band of the oxide with the highest occupied molecular orbital of the adsorbate and the highest energy states of the valence band and the lowest unoccupied molecular orbital for O2, respectively. This provides for the best overlap that would permit charge transfer and reactivity[32]. The band gap also relates to
the reducibility of the oxide, which in some cases can correlate with both the activity and the selectivity[35]. The band gap of oxide can, in principle, be tuned by changing
the domain size of the active surface ensembles or by changing the properties of the metal oxide support.
3) Oxidation state of the metal ion.
As in organometallic chemistry, the oxidation state of the metal ion can have a profound e ect on its ultimate reactivity. The oxidation state strongly influences the redox and acid–base properties of the metal ion center[35] . Since most elementary reactions intimately involve the metal, changes in its oxidation state will strongly control its ability to carry out specific reaction steps.
4) Acid–base properties available at di erent adsorption sites.
The Lewis and Brønsted acid and base properties dictate whether a molecule will adsorb at a particular site along with the specific reaction chemistry that may occur at that site. Many probe molecules have been used to try to help identify the acid and basic characteristics for particular reactions. The best probe molecule is one which specifically tests for the specific reaction of interest. The strength best adsorption probe for acid and base site strength will clearly depend on the degree of acidity or basicity needed for the specific reaction. By using isopropanol as a probe molecule to characterize both acid and base sites, Kulkarni and Wachs[33] showed that the turnover frequency for isopropanol oxidation changed by over eight orders of
magnitude with changes in the metal oxide. The surface acidity is therefore a highly tunable property[36] that may be used to control reactivity. Macht et al.[[37] used 2,6-di-tert-butylpyridine as a selective probe of the Brønsted acid sites that form in situ via the dehydration of butanol over supported WOx complexes. Their results demonstrate strong support e ects on the overall rate. The intrinsic turnover frequency per acid site for butanol dehydration, however, remained relatively constant. The acid site density was found to be strongly tied to the nature of the support. They showed a general correlation between the Sanderson electronegativity of the cation in the support and the measured Brønsted acid regioselectivities.
5) Chemical bonding in oxides: covalent vs. ionic
The nature of the bonds that form within the oxide control its oxidation state and sets its ability to participate in carrying out specific reactions. In addition, the bond types tend to control the mechanisms by which reactions proceed. These e ects can either be direct or occur more subtly. Some oxides actually take on both covalent as well as ionic characteristics, as was discussed in Section 5.3[34].
6) Defect sites.
Various defects are known to form within the surface of an oxide including the presence of anion vacancies (F-centers), cation vacancies, interstitials, trapped electrons, or holes.[35] These defects change the local electronic structure and can significantly
Catalysis by Oxides and Sulfides 241
impact surface reactivity[29,32,38−40]. The surface chemistry for a range of di erent systems may actually occur at defect sites since they expose coordinatively unsaturated centers.
In Chapter 2, we indicated that some of the most viable routes for the selective oxidation of propane to acrylonitrile today appear to come from substituted vanandium antimonates (VSbO). There have been various studies carried out to understand the influence of cation substitution. Andersson et al. [41] demonstrated increased catalytic performance for acrylonitrile production when Ti was substituted into VSbO oxides. They suggested that the increased performance was due to the prevention of V2O5 formation and the stabilization of site-isolated vanadium. Xiong et al.[42] followed up on these ideas in an elegant study in which they combined UV–Raman spectroscopy, X-ray di raction and periodic DFT calculations to elucidate the influence of Ti on vanadium antimony oxide and its redox behavior. The combination of UV spectroscopy and DFT frequency calculations was used in this work to identify the bulk rutile structure, the characteristic modes of V2O5 and the formation of unique bands at 880 and 1016 cm−1. Theory together with experiment was used to identify these bands as stretching bands for two-coordinate oxygen species which reside at SbOSb bridges (880 cm−1) and SbOV bridges (1016 cm−1)that sit near a cation vacancy. Oxygen, in the rutile structure, is predominantly surrounded by three cations and is thus considered three-coordinate. The twocoordinate oxygen sites are the result of oxygen adsorption near the cation vacancies. Exposure of the surface to ammonia shows the complete removal of these bands, which suggests that ammonia is predominantly activated at these sites to produce water. These bands reappear, however, when the sample is exposed to air, which indicates that they are readily reoxidized. The incorporation of Ti into the vanadium antimonate structure was found to enhance the number of sites as measured by the increased intensity of the UV band and by the ease of formation. This suggests that Ti appears to improve redox properties. The authors speculate that ammonia reacts with the bridging oxygen species at the SbOSb sites, thus removing water and forming SbNHSb sites, which take part in the ammoxidation of the hydrocarbon. They speculate that these Sb sites are active in the formation of acrylonitrile. The substitutional addition of Ti was also found to help stabilize V3+ and V4+ cations and hence prevent the breakdown of vanandium antimonate into V2O5 surface structures.
Other factors which are also important include the following:
7) Surface morphology and structure.
Metal oxides can demonstrate remarkably di erent behavior for reactions carried out over di erent surfaces. The phase, morphology and the exposed surface facets can all be important. For many reaction systems there are well-prescribed phases and crystal faces which appear to dominate activity and/or selectivity. For example, the active phase for butane oxidation to maleic anhydride is thought to be (VO)2P2O7. The (100) surface for this system appears to be the most active. The nature of both the oxygen and the vanadium cations in VPO can be quite di erent depending upon which surface is exposed[29,40]. In addition, the surface structure itself can be quite di erent depending on the nature of the support. The surface structure can dramatically change as the reaction proceeds and conditions change.
242 Chapter 5
8) Surface termination.
Depending upon the reaction conditions, the active surface can take on various di erent forms. Under highly oxidizing conditions, most of the sites may be covered by oxygen. More reducing conditions, on the other hand, will likely expose coordinatively unsaturated sites that take on more metallic-like properties. Intermediate conditions can give rise to interesting interfaces between oxide and metal-like configurations[44]. Most of the theoretical, and surface science studies, however, have been carried out over model surface structures which may potentially oversimplify the true nature of the surface. Fundamental studies have looked at both neutraland polar-terminated surfaces and demonstrate that these two surfaces can lead to rather di erent kinetics[44,45].
9) Site isolation and domain size.
Under the conditions of interest, a number of mixed metal oxides may exist as siteisolated metal oxide complexes or as two-dimensional chains that wet the surface of the support. The nature of the bridging metal–oxygen–support bonds can ultimately begin to control the surface chemistry. The optimal active domain size strongly
depends upon the reaction of interest. Wachs and co-workers[46,47] and Corma and Garcia[36] have shown that isolated MOx sites are selective and, in some cases, more
active in carrying out di erent selective oxidation reactions. Macht et al. [37] have demonstrated that the optimal supported-WOx domains for alcohol dehydration are two-dimensional domains of size 9–10 W/nm2.
10) Influence of water.
Water can play an important role in changing the surface composition, thus leading to an increase in the number of surface hydroxyl intermediates as well as Brønsted acid sites. This can enhance the kinetics for various di erent reactions[33,48−50]. In other instances, water can act to block sites and impede surface kinetics.
11) Metal oxide support and ligand e ects.
The metal–oxygen bonds as well as the properties of the oxygen can change significantly if they are attached to a second metal such as in the M–O–S or M–O–M’ systems where S refers to the support and M’ refers to the second metal. Wachs
and co-workers, for example, have shown that there are significant changes in activity with changes in the metal oxide support[33,48−50]. As such, the support can be
thought of as a potential ligand which can be tuned to tailor the activity and selectivity of the molecular oxide surface layer. Wang and Wachs demonstrate a 12-fold
increase in the methanol decomposition rate over V2O5 supported on silica, alumina, titania, and ceria[50]. The origin of the increase in activity is thought to be due to the
ligand e ect whereby the changes can be correlated with the electronegativity of the cation that corresponds to the ligand. The more electropositive cations such as Ti and Ce have substantially higher turnover frequencies than the more electronegative Al and Si cations.
In the following sections, we elaborate in more detail on how the structural and electronic properties of the oxide control its catalytic behavior by following a few example systems. We describe the applications to oxidation chemistry over reducible and non-reducible oxides, and acid chemistry via specific example catalytic systems.
Catalysis by Oxides and Sulfides 243
5.6.2 Lessons Learned from Surface Science
Before discussing the link between the properties and performance of supported metal oxide particles, we first review how some of the factors discussed above control the chemistry on well-defined metal oxide surfaces. Surface science has provided a wealth of information which has been used to advance our understanding of the elementary processes that occur on transition metal surfaces and the factors that control them. This was discussed in detail in Chapters 2 and 3. Our understanding of metal oxides, however, is much less mature. This is due to the complexity of the structure and properties of metal oxides and the di culty of carrying out well-defined UHV experiments that are not masked by the complexity of the system. More recent e orts have shown that many of these di culties can be overcome. This has helped to establish much of the current interest in this area. While the fundamental surface science on metal oxides is a less mature subject, it this has become a topic of much greater interest over the past decade. The interested reader is pointed to elegant reviews on the fundamental properties of single-crystal metal oxide surfaces by Heinrich and Cox[51], Freund[43], and Barteau[35] .
Here we follow the elegant analysis presented by Barteau [35] as it discusses some of the critical factors that control the activity and selectivity of metal oxides. In addition, it illustrates how the surface structure and properties can be controlled even under reducing UHV conditions in order to impart specific reactivity.
The surface structure and properties can be modulated in order to establish specific surface ensembles active for carrying out a range of di erent oxygenate and hydrocarbon coupling reactions. However, by specifically controlling
1)the coordination number of the surface cations
2)the oxidation states of the cations
3)the redox properties of a well-defined oxide surface
Barteau[35,52] was able to demonstrate for well-defined TiO2 and ZnO surfaces the activity and selectivity for C2 oxygenate (carboxylates and aldehydes) and hydrocarbon (alkynes) coupling reactions over these model metal oxide surfaces under UHV conditions that is typically only seen in organometallic systems in solution.
They found that three di erent C–C bond formation paths could occur over di erent TiO2 surfaces depending upon the nature of the surface, its properties and specific properties of these surfaces as well as the reaction conditions. The reaction paths identified included carboxylate ketonization, base-catalyzed aldol condenstation and reductive carbonyl coupling. Each reaction was found to be sensitive to the specific electronic and structural properties of the cation sites available at the surface of the oxide as well as the nature of the sites local environment. Carboxylate coupling or ketonization was shown to occur most favorably over the TiO2 (001)-[114] faceted surface which contains a distribution of 4-, 5-, and 6-coordinate Ti sites. The high number of the coordinatively unsaturated Ti sites were thought to be responsible for the higher selectivity of the bimolecular coupling of acetate groups over their unimolecular decomposition paths. This is a stable surface that is formed at higher temperatures. Cation sites which contain pairs of vacancies were thought to be necessary in the active ensemble in order to accommodate the electron pairs that result from the coadsorption of the two ligands to the same center.
Aldol condensation, on the other hand, is favorably carried out over basic oxygen sites on the TiO2 surface and does not appear to require coordinatively unsaturated Ti sites. The reaction is thought to proceed initially by the proton abstraction at the carbonyl carbon from the aldehyde reagent molecule by a basic oxygen site on the surface
244 Chapter 5
and the subsequent formation of an adsorbed enolate intermediate. This is followed by a nucleophilic attack on the adsorbed enolate by a second aldehyde reactant molecule. The coupled intermediate that forms then undergoes a dehydration in order to from the αβ-unsaturated aldehyde product. This occurs much more like that of an “outer-sphere” mechanism typically found in homogeneous solution-phase catalysis (discussed in more detail in Chapter 6) whereby there is a reaction between the adsorbed enolate ion and the gas phase reagent molecule (ion–molecule reaction). The reaction does not require the initial formation of a bimolecular surface ensemble. The carboxylate ketonization reaction discussed early, however, requires the explicit reaction between two surface intermediates (ion–ion reaction). This reaction they can therefore be considered to follow more of an “inner-sphere” mechanism.
The aldol condensation reaction demonstrates conversions on the order of 60% and selectivities to coupling products of 96% when carried out over the TiO2 (001)-[114] surface. The yield is increased even further on going to the [001] faceted TiO2 (001)- [001] surface and thus approaches those found over TiO2 powders (run under similar conditions). The patent literature shows yields that are only slightly higher with 80% conversion and selectivities of 90%.
The third and final coupling path involves the reductive coupling of two carbonyl groups which requires the ability to facilitate a four-electron reduction process. Redox now is much more important. This reaction therefore requires the presence of reduced lower valent cations. This can be established, however, by having ensembles of lower valent cations. The mechanism is thought to involve the reductive coupling of two aldehyde reagents to form a diolate, also known as the pinacolate intermediate, which is bound to the surface through its two oxygen atoms. This intermediate subsequently dissociates to form an alkene leaving behind two oxygen atoms at the surface. XPS studies clearly reveal the presence of Ti+, Ti+2 and Ti+3 oxidation states and their importance in the reaction chemistry.
5.6.3 Redox Considerations
Haber and Witko[32] developed a simple picture of redox processes based on frontier orbital concepts that were presented in Chapter 3 in order to begin to assess metal-oxide bonding and the ability of the oxide to carry out redox chemistry. Many oxidation reactions are carried out via a Mars–van Krevelen-like mechanism, which involves the activation of an organic molecule (RH) and the subsequent insertion of either a surface oxygen atom or lattice oxygen into the hydrocarbon surface intermediate. Oxygen at the surface is then replenished by the activation of O2. This process requires that cations in oxide be able to readily change oxidation states. This can be described quite simply by the following redox couple:
RH + O2− −→ R−O− + H+ + 2e− |
(5.2a) |
|
1 |
O2 + 2e− −→ O2− |
(5.2b) |
2 |
||
In order for the redox cycle depicted in Eqs. (5.2a) and (5.2b) above to proceed spontaneously, two conditions must be satisfied. First, the highest molecular orbital of the organic molecule must be above the Fermi level and aligned with electronic states at the
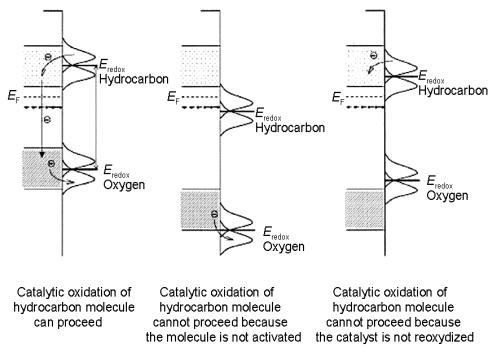
Catalysis by Oxides and Sulfides 245
Figure 5.17. Schematic comparisons of the di erent electronic interactions between a gas-phase hydrocarbon molecule, oxygen and an oxide surface. A necessary, but not su cient, requirement for a redox reaction to occur is that the hydrocarbon can donate electron density into the bottom of the conduction band which lies above the Fermi level whereas electron density can be transferred from the top of the valence band into splitting O2 at the surface. This requires the appropriate alignment of the highest occupied orbitals from the hydrocarbon with the bottom of the conduction band and the lowest unoccupied orbitals of O2 with the top of the valence band as in the schematic on the left. The inappropriate alignment of states is shown in the scheme at the center and in the scheme on the right hand side, both of which would prevent reaction from occurring[32].
bottom of the unoccupied conduction band so that electron density can be transferred from the organic reactant to the surface states. The second condition is that the unoccupied molecular orbitals of the O2 molecule must sit below the Fermi level in line with occupied states in the valence band. The appropriate alignment is shown in the schematic on the left hand side of Fig. 5.17. The alignment of the states in the other two schematics is such that electron transfer will not proceed. The potentials of the conduction band can be changed by the creation of:
(a)an interface with a second metal oxide surface,
(b)addition of di erent valence ions,
(c)formation of defect sites, or an applied potential.
These ideas present a simple picture of how one can influence the reactivity in redoxbased systems. While the picture is somewhat oversimplified, it o ers a useful tool for conceptually understanding the reactivity of redox systems.