

Pat_fiziologiya_Ataman
.pdfзменшенні кількості тромбоцитів нижче 50 109/л.
За походженням тромбоцитопенії можуть бути спадково обумовленими і набутими. За механізмом розвитку виділяють такі види тромбоцитопенії. І.
Тромбоцитопенії, пов'язані з порушеннями утворення тромбоцитів:
а) мієлотоксичні тромбоцитопенії— виникають унаслідок ушкодження кровотворних клітин. Дуже часто поєднуються з анемією й лейкопенією. Причинами їх розвитку є ті самі фактори, які викликають розвиток гіпопластич-ної анемії; б) дефіцитні тромбоцитопенії— обумовлені недостатністю вітаміну В12 або
фолієвої кислоти; в) дисрегуляторні тромбоцитопенії— пов'язані з порушенням утворення
тромбоцитопоетинів — речовин, що стимулюють утворення тромбоцитів;
г) тромбоцитопенії, пов'язані зі зменшенням плацдарму кровотворення, —
розвиваються при лейкозах і метастазах злоякісних пухлин.
її. Тромбоцитопенії, пов'язані з посиленим руйнуванням тромбоцитів. Причиною такого руйнування можуть бути:
а) імунне ушкодження, обумовлене антитромбоцитарними антитілами на власні компоненти кров'яних пластинок або на лікарські препарати, адсорбовані на тромбоцитах. Аутоімунне ушкодження вважають найбільш імовірним механізмом розвитку так званої ідіопатичноїтромбоцитопенічноїпурпури (хвороби Верльгофа);
б) ггперспленізм — гіперфункція селезінки, що супроводжується часто спленомегалією. У результаті підвищення фагоцитарної активності макрофагів відбувається інтенсивне руйнування всіх формених елементів крові, у тому числі й тромбоцитів; в) механічне ушкодження тромбоцитів. Часто виникає при гемангіомах і
накладенні штучних клапанів серця; г) набутімембранопатії(гемолітична анемія Маркіафави—Мікеллі). Соматичні
мутації кровотворних клітин спричиняються до утворення пулів клітин (еритроцитів, гранулоцитів, тромбоцитів) з дефектами мембрани. У результаті збільшується чутливість таких клітин до дії комплементу й відбувається їх руйнування. III. Тромбоцитопенії споживання. Виникають у результаті посиленого використання тромбоцитів на утворення тромбів (хвороба Шенляйн—Геноха, хвороба Мошковича, ДВЗ-синдром).
26.3.16. Який патогенез порушень гемостазу в умовах тромбоцитопенії?
У патогенезі геморагічного синдрому при тромбоцитопеніях мають значення:
1)порушення ангіотрофічної функції тромбоцитів, у результаті чого виникають дистрофічні зміни в ендотелії і збільшується ламкість мікросудин. Це веде до збільшення ранимості судин, діапедезу еритроцитів, крововиливів. Останні виявляють себе петехіями на шкірі, кровотечами з ясен і носа, крововиливами в головний мозок і сітківку ока;
2)порушення адгезії й агрегації тромбоцитів. Це викликає порушення формування тромбоцитарного тромбу й призводить до збільшення часу кровотечі (проба Дюка);
3)порушення вторинного спазму ушкоджених артеріол. При тромбоцитопеніях вивільняється мало біогенних амінів (катехоламінів, серотоніну), з дією яких пов'язане скорочення гладких м'язів судин;
4)порушення зсідання крові. Обумовлено недостатнім вивільненням фактора 3
пластинок і тромбостеніну. У результаті порушується І фаза зсідання крові і ретракція згустку.
26.3.17.Що таке тромбоцитопатії? Як їх класифікують?
Тромбоцитопатії—це порушення функціональних властивостей тромбоцитів, йґ якісна неповноцінність. При цьому кількість тромбоцитів може залишатися в нормі
J
За походженням тромбоцитопатії бувають спадково обумовленими і набутими.
За характером якісних дефектів кров'яних пластинок тромбоцитопатії поділяють на
ендо- і екзотромбоцитарні.
Ендотромбоцитарні тромбоцитопатії обумовлені порушеннями складових частин тромбоцитів. їх, у свою чергу, поділяють на мембранопатії, гранулопатії і ферментопатії. Мембранопатії виникають при спадкових аномаліях мембранних пгіко-протеїнів, що виконують функції клітинних рецепторів; при блокаді цих рецепторів аномальними білками плазми крові (парапротеїнами), при ушкодженні мембрани кров'яних пластинок патогенними факторами. Гранулопатії виявляються дефіцитом гранул І і II типів. В основі ферментопатії може лежати зменшення активності ферментів циклу Кребса, гліколізу, порушення функцій АТФ-аз, циклоксигенази і тром-боксансинтетази.
При екзотромбоцитарних тромбоцитопатіях причини порушення функцій тромбоцитів лежать поза кров'яними пластинками. У зв'язку з цим екзотромбоцитарні тромбоцитопатії можуть бути:
а) пов'язаними зі змінами плазми крові (дефіцит плазмових білків, що є плазмовими кофакторами агрегації тромбоцитів); б) пов'язаними зі змінами в судинній стінці (порушення утворення фактора Вілле-
бранда ендотелієм судин, розлади зовнішнього механізму зсідання крові). Залежно від сутності порушень гемостазу виділяють:
1) тромбоцитопатії з первинним порушенням адгезії тромбоцитів^
2) тромбоцитопатії з первинними порушеннями агрегації тромбоцитів;
3) тромбоцитопатії з первинним порушенням реакцій вивільнення вмісту тромбоцитів',
4) тромбоцитопатії, пов'язані з дефіцитом або зменшенням доступності фактора З тромбоцитів.
26.3.18.Наведіть приклади тромбоцитопатій з різними механізмами порушень гемостазу.
1. Тромбоцитопатії з первинним порушенням адгезії тромбоцитів:
а) хвороба Віллебранда — ангіогемофілія. Обумовлена генетичними порушеннями синтезу фактора Віллебранда ендотеліальними клітинами (тип спадкування аутосомно-домінантний);
б) хвороба Бернара—Сульє — макротромбоцитодистрофія. Причиною є спадково обумовлений дефект глікопротеїнів тромбоцитарної мембрани (ГШЬ), що взаємодіють з фактором Віллебранда. При цьому тромбоцити набувають гігантських розмірів. Тип спадкування аутосомно-рецесивний.
2. Тромбоцитопатії з первинними порушеннями агрегації тромбоцитів - дизагрегаційні. Найчастіше буває тромбастенія Гланцмана, що виникає як наслідок дефектів мембранних глікопротеїнів І і II типів, що беруть участь в агрегації. При цьому адгезія тромбоцитів і вивільнення їхніх гранул відбуваються, а агрегація -ні, незважаючи на дію таких потужних агрегантів, як АДФ, адреналін, тромбін. Тип спадкування аутосомно-рецесивний.
3.Тромбоцитопатії з первинним порушенням реакцій вивільнення вмісту тромбоцитів:
а) порушення дегрануляції тромбоцитів — "парез реакції вивільнення ". Виникає,
зокрема, при порушенні утворення тромбоксану А2 при дії ацетилсаліцилової кислоти. Показано, одо одноразове приймання аспірину необоротно збільшує час вивільнення гранул від 3,5 до 6 діб, поки не з'являться нові тромбоцити; б) недостатність накопичення й збереження вмісту гранул тромбоцитів. Виникає, як правило, при генетично обумовлених порушеннях.
Розлади реакцій вивільнення гранул викликають порушення другої хвилі агрегації тромбоцитів. Початкова агрегація кров'яних пластинок закінчується їх дезагрегацією.
4.Тромбоцитопатії, пов'язані з дефіцитом або зменшенням доступності фактора 3 пластинок. У їхній основі можуть лежати або генетично обумовлені дефекти структури цього фактора, або порушення його вивільнення з ушкоджених тромбоцитів. При цьому порушується зсідання крові (коагуляційний гемостаз). Адгезив-но-агрегаційні властивості тромбоцитів не міняються.
26.3.19.Чим можуть бути обумовлені порушення коагуляційного гемостазу - коагулопатії?
В основі розвитку коагулопатій можуть бути:
1) зменшення активності системи зсідання крові;
2) підвищенням активності антикоагулянтної системи;
3) збільшенням активності фібринолітичної системи.
26.3.20.Що може бути причиною безпосереднього порушення І фази зсідання крові?
Залежно від характеру порушень І фази зсідання крові виділяють три групи розладів.
I. Ізольовані порушення зовнішнього механізму активації зсідання. Виникають при дефіциті ф. VII - гіпопроконвертинемії. Цей дефіцит може бути спадково обумовленим (тип спадкування аутосомно-рецесивний) або набутим (гіповітаміноз К, ураження печінки).
II. Ізольовані порушення внутрішнього механізму активації зсідання:
а) дефіцит ф.УШгемофілія А. Найчастіше виникає як генетичний дефект коагулянтної частини ф.УШ (тип спадкування зчеплений з Х-хромосомою). Можливе утворення аутоантитіл проти білкових компонентів цього фактора зсідання;
б) дефіцит ф. IXгемофілія В. Причиною розвитку є спадкова патологія (тип спадкування зчеплений з Х-хромосомою), дефіцит вітаміну К або ураження печінки, антитіла проти ф. IX;
в) дефіцит ф. XI— гемофілія С. Виникає при генетичних порушеннях (тип спадкування аутосомно-рецесивний) або ураженнях печінки; г) дефіцит ф. ХП. Спадкова патологія, що буває дуже рідко. Завдяки калікре-їн-
кініновій системі цей дефект добре компенсується, оскільки запуск внутрішнього механізму зсідання відбувається через зовнішній; ґ) дефіцит ф. З тромбоцитів. Є наслідком тромбоцитопенії або певних видів тромбоцитопатій.
III. Поєднані порушення зовнішнього і внутрішнього механізмів зсідання. Розвиваються при дефіциті ф. X, що може бути спадково обумовленим (тип спадкування аутосомно-рецесивний) або набутим (гіповітаміноз К, ураження
печінки).
26.3.21.Що може бути причиною безпосереднього порушення II фази зсідання крові?
1. Дефіцит ф.ІІ— гіпопротромбінемія. Найчастіше має набутий характер і розвивається внаслідок гіповітамінозу К або уражень печінки.
2. Дефіцит ф. V— парагемофілія. Порушення утворення проакцелерину може бути обумовлено ураженнями печінки або аутоантитілами проти ф.У
26.3.22.Що може бути причиною безпосереднього порушення III фази зсідання крові?
1. Дефіцит фібриногену:
а) афібриногенемія - повна відсутність фібриногену (спадкове захворювання з аутосомно-рецесивним типом спадкування); б) гіпофібрииогенемія - зменшення синтезу фібриногену в печінці при її ураженнях.
2. Дисфібршшгенемії - якісні порушення фібриногену. Розвиваються як наслідок генетичних дефектів (тип спадкування аутосомно-домінантний). Виявляються утворенням аномального фібрину.
3.Порушення полімеризації фібрину. Розвиваються Б результаті утворення комплексів фібриногену з фібрином-мономером і проміжними продуктами, від яких ще повністю не відщепились пептиди А і В. При цьому утворюється так званий "заблокований фібриноген" ("тромбінрезистентний фібриноген"), що не піддається дії тромбіну.
4.Дефіцит ф. ХШ. Виникає як наслідок спадкових порушень (тип спадкування ау- тосомно-рецесивний). виявляється порушеннями перетворення розчинного фібрину (фібрину S) у нерозчинний (фібрин І).
26.3.23.Які речовини складають антикоатулянтну систему крові?
І. Первинні антикоагулянти. Постійно синтезуються в організмі й тому завжди містяться в плазмі крові. До них відносять:
1) антитромбін ПІ- основний універсальний антикоагулянт, що є інгібітором протеаз. Синтезується ендотелієм судин. Пригнічує активність усіх протеолітичних ферментів крові, у тому числі тромбіну, калікреїну, плазміну, ф. ХІІа, ф. ХІа, ф. Ха, ф. ІХа, ф.УІІа;
2) гепарин (антитромбін II) — глікозаміноглікан, що вивільняється тканинними базофілами і базофілами крові при їх дегрануляції. Антикоагулянтні властивості має не сам гепарин, а комплекс гепарину з антитромбіном III. Завдяки гепарину антитромбін III фіксується на поверхні ендотелію судин, де його антикоагулянтні властивості в багато разів зростають;
3) а -антитрипсин, а2-макроглобулін, інгібітор СІ-компонента комплементу - усі вони є неспецифічними інгібіторами протеаз, у тому числі й факторів зсідання крові.
її. Вторинні антикоагулянти. У плазмі крові в нормі не містяться. Утворюються в процесі зсідання крові й фібринолізу. До них відносять:
1) антитромбін І— фібрин, що адсорбує і в такий спосіб інактивує велику кількість тромбіну; 2) продукти фібринолізу. Перешкоджають процесам полімеризації фібрину і
утворенню фібринових структур.
26.3.24.ЧИМ може бути обумовлене підвищення активності антикоагулянтної системи крові?
Підвищення активності антикоагулянтної системи крові закономірно виникає при:
1)збільшенні вмісту гепарину в крові — гіпергепаринеміг. Це може бути обумовлено посиленою дегрануляцією тканинних базофілів і базофілів крові, зокрема, при алергічних реакціях І типу за класифікацією Кумбса і Джелла, руйнуванням базо-фільних лейкоцитів при лейкозах, введенням гепарину ззовні;
2)появою "патологічних " антикоагулянтів, до яких відносять антитромбін V, що порушує полімеризацію фібрину-мономеру; "вовчаковий" антикоагулянт, що порушує утворення протромбіназного комплексу; парапротеїни, що унеможливлюють полімеризацію фібрину.
26.3.25. Що ВХОДИТЬ у ПОНЯТТЯ "фібринолітична система"?
Фібриноліпшчна система — це система, яка забезпечує лізис (протеоліз) фібрину в кровоносному руслі. У такий спосіб вона бере участь у підтримці рідкого стану крові й у відновленні кровообігу в тромбованих судинах.
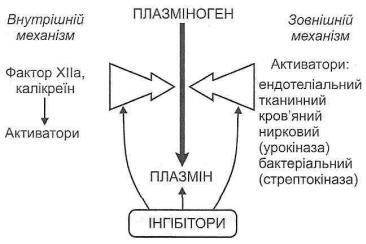
До складу системи фібринолізу входять (рис. 119):
Рис. 119. Схема фібринолізу
1)плазміноген (профібринолізин) — неактивний протеолітичний фермент, що завжди міститься в плазмі крові;
2)плазмін (фібринолізин) — активна форма плазміногену. Утворюється в результаті дії активних протеаз на плазміноген і відщеплення від його молекули пептиду, який "закриває" активний центр;
3)активатори фібринолізу — велика група речовин, які або самі є протеазами і здатні перетворювати плазміноген у плазмін, або викликають появу таких протеаз;
4)інгібітори фібринолізу. До них відносять інгібітори протеаз, серед яких
найбільше значення має а2-антиплазмін.
Розрізняють внутрішній і зовнішній механізми активації фібринолізу. Внутрішній механізм обумовлений активацією фактора XII зсідання крові й утворенням калікреїну, унаслідок чого в крові з'являється велика кількість активаторів фібринолізу.
Зовнішній механізм пов'язаний з надходженням у кров готових активаторів фібринолізу: ендотеліального, клітинного, тканинного (урокіназа), бактеріального (стрептокіназа).
26.3.26. Які фактори викликають підвищення активності фібриполітичної системи крові?
1. Посилене утворення й надходження в кров активаторів фібринолізу.
Відбувається при великих ушкодженнях тканин: великі травми, ушкодження клітин токсинами, операційні втручання, лейкози та ін.
2. Зменшення вмісту в крові інгібіторів протеолізу. Має місце при недостатньому їх утворенні або посиленому використанні.
26.3.27.ЯКИМИ КЛІНІЧНИМИ ознаками виявляють себе порушення коагуляційного гемостазу?
На відміну від порушень судинно-тромбоцитарного гемостазу для коагулопатій характерні не капілярні (точкові) кровотечі, а кровотечі з великих судин — артерій і вен. Такі кровотечі клінічно виявляють себе:
а) гематомами — великими крововиливами в м'язи, під шкіру, у порожнину суглобів (гемартрози); б) тривалими кровотечами після операційних втручань (видалення зуба та ін.).
26.3.28.Що таке ДВЗ-синдром?
Синдром дисемінованого внутрішньосудинного зсідання крові (ДВЗ-син-дром) — це генералізоване зсідання крові всередині судин, що викликає утворення великої кількості мікрозгустків і агрегатів клітин, які порушують мікроциркуляцію в органах і тканинах.
Цей синдром часто характеризують як катастрофу для організму.
26.3.29. Що може бути причиною ДВЗ-синдрому?
Залежно від причин розвитку виділяють такі різновиди ДВЗ-синдрому:
1)інфекційно-септичний (розвивається при сепсисі);
2)посттравматичний (при краш-синдромі, опіковій хворобі, множинних переломах кісток);
3)шокогенний (при всіх видах шоку);
4)хірургічний (після операцій з великою травматизацією тканин);
5)акушерський (при передчасному відшаруванні плаценти, надходженні в кров навколоплідних вод);
6)токсигенний (після укусу змії);
7)пухлинний (при злоякісному пухлинному рості);
8)алергічний (при імунному ушкодженні тканин) та ін.
26.3.30. Що є патогенетичною основою розвитку ДВЗ-синдрому?
В основі патогенезу ДВЗ-синдрому лежить так званий "гуморальний протеазний вибух ", тобто одночасна активація всіх протеолітичних ферментів плазми крові, що входять до складу чотирьох позаклітинних біохімічних систем (рис. 120):
а) системи зсідання крові; б) фібринолітичної системи;
в) калікреїн-кінінової системи; г) системи комплементу.
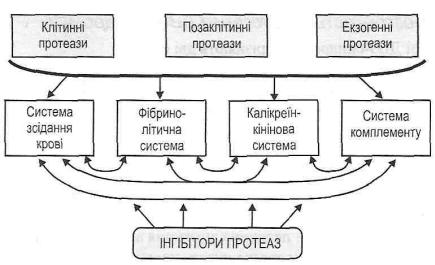
Рис. 120. "Гуморальний протеазний вибух"
Основний принцип активації позаклітинних протеаз — відщеплення пептидів, що закривають їхні активні центри. Утворення активних протеолітичних ферментів крові має свої особливості:
а) можлива самоактивація ферментів — активний фермент, впливаючи на неактивну форму, переводить її в активну; б) одні активні протеази здатні активувати інші (перехресна активація)',
в) ланцюговий характер активації. Теоретично поява навіть однієї молекули активної протеази може викликати активацію всіх наявних протеаз крові. Однак у нормі реакції активації протеолітичних ферментів мають обмежений характер, що пояснюється існуванням великої групи інгібіторів протеаз.
При патології, коли у кров надходять великі кількості активних протеаз, потужність існуючих інгібіторів може виявитися недостатньою. Отоді й виявить себе ланцюговий характер активації протеолітичних систем плазми крові. Така активація набуває генералізованого характеру, втягує всі протеази крові - відбувається "гуморальний протеазний вибух".
26.3.31. Назвіть основні джерела надходження в кров активних протеаз при ДВЗсиндромі.
Існує три основних джерела надходження протеаз у кров.
I. Ушкоджені клітини. Має значення гостре ушкодження великої кількості клітин, з яких у позаклітинний простір і кров надходять лізосомні протеази, тканинний тромбопластин.
Запалення як місцевий процес, що виникає при ушкодженні клітин, обмежує надходження продуктів розпаду в кров, локалізуючи ушкодження і в такий спосіб попереджаючи розвиток ДВЗ-синдрому.
II. Надходження в кров великої кількості позаклітинних протеаз; наприклад трипсину при гострому панкреатиті, ферментів, що містяться в навколоплідних водах.
III. Екзогенні протеази. їхніми джерелами можуть бути бактеріальні клітини при сепсисі, зміїна отрута та ін.
26.3.32. Як розгортається патогенез ДВЗ-синдрому?
У патогенезі ДВЗ-синдрому розрізняють дві фази.
I фаза - фаза гіперкоагуляції і агрегації тромбоцитів. Основу цієї фази становить генералізована активація системи зсідання крові, тобто утворення тромбіну (тромбінемія), що призводить до утворення фібрину і агрегатів тромбоцитів.
Існує три механізми запуску цієї фази:
1)ферментативний механізм - надходження в кров великої кількості активних протеаз і тканинного тромбопластину;
2)контактний механізм - активація ф. XII при контакті його з чужорідними поверхнями (екстракорпоральний кровообіг, гемодіаліз, штучні клапани серця);
3)тромбоцитарний механізм - первинна активація агрегації тромбоцитів при гене-ралізованому ушкодженні ендотелію судин, порушеннях реологічних властивостей крові, гострому внутрішньосудинному гемолізі еритроцитів.
У результаті реалізації зазначених механізмів утворюється велика кількість мікрозгустків і агрегатів клітин, що призводить до розладів мікроциркуляції, розвитку сладж-синдрому (див. розд. 13).
Усе це веде до появи таких клінічних проявів, як гіпоксія, ацидоз, інтоксикація продуктами розпаду, гостра недостатність зовнішнього дихання (мікрозгустками закупорюються капіляри легень), гостра ниркова недостатність (забиваються капіляри клубочків), порушення мозкового кровообігу.
II фаза - фаза гіпокоагуляції (геморагічний синдром). Ця фаза розвивається як наслідок виснаження механізмів судинно-тромбоцитарного і коагуляційного гемостазу.
У її виникненні мають значення:
а) зменшення активності системи зсідання крові (споживання факторів І, V, VIII); б) активація фібринолітичної системи (надходження в кров великої кількості активаторів фібринолізу); в) підвищення антикоагулянтної активності крові за рахунок утворення продуктів фібринолізу;
г) розвиток тромбоцитопенії споживання; ґ) підвищення проникності стінки судин (має значення утворення великих
кількостей кінінів). Фаза гіпокоагуляції клінічно виявляє себе масивними кровотечами, що їх важко зупинити.
26.3.33. Що таке тромбофільні діатези? Що може лежати в основі їх розвитку?
Тромбофільні діатези - це захворювання і синдроми, при яких схильність до утворення тромбів обумовлена первинними порушеннями механізмів гемостазу. Патогенетичну основу тромбофілій можуть становити: 1) ендотеліальні механізми, що обумовлюють зменшення тромборезистентності стінки судин;
2)тромбоцитарнімеханізми, пов'язані зі збільшенням агрегаційної здатності тромбоцитів;
3)зменшення антикоагулянтної активності крові (зменшення утворення антитромбіну III);
4)зниження активності фібринолітичної системи (зменшення утворення ендоте-
ліального активатора плазміногену, поява потужних інгібіторів плазміну, дефіцит плазміногену);
5)порушення системи зсідання крові (дисфібриногенемії).
Клінічно тромбофільні діатези виявляють себе тромбозами і тромбоемболіями венозних і артеріальних судин у різних органах і тканинах.
27. Патологічна фізіологія серця
27.1. Що таке недостатність кровообігу? Чим вона може бути
обумовлена?
Недостатність кровообігу - це стан, при якому серцево-судинна система не може забезпечити органи й тканини організму необхідною кількістю крові. Є найчастішим проявом різних порушень функцій системи кровообігу. Недостатність кровообігу може бути обумовлена:
1)недостатністю серця;
2)недостатністю кровоносних судин;
3)серцево-судинною недостатністю, тобто одночасною недостатністю серця і судин.
27.2. Які наслідки для органів, тканин і організму в цілому має недостатність кровообігу?
1. Порушення трофічного забезпечення органів і тканин. Розвиваються як наслідок:
а) зменшення доставки кисню (циркуляторпа гіпоксія, див. розд. 19); б) зменшення доставки поживних речовин.
2. Порушення видалення з органів і тканин кінцевих продуктів обміну речовин.
Наслідком цього є розвиток:
а) негазового ацидозу; б) інтоксикації.
27.3. Що таке недостатність серця?
Недостатність серця - це патологічний стан, обумовлений нездатністю серця забезпечити кровопостачання органів і тканин відповідно до їхніх потреб.
Це стан, при якому навантаження на серце перевищує його здатність виконувати роботу. Він виявляє себе тим, що серце не здатне переміщати в артеріальне русло всю кров, що надходить до нього по венах.
27.4. Як класифікують недостатність серця?
I. Залежно від клінічного перебігу розрізняють гостру і хронічну недостатність серця.
II. За вираженістю клінічних проявів недостатність серця може бути прихованою
(компенсованою) і явною (декомпенсованою).
III. Залежно від порушення функції переважно того чи того відділу серця
розрізняють лівоишуночкову, правошлуночкову і тотальну недостатність серця.
IV. За патогенезом виділяють:
а) недостатність серця від перевантаження; б) міокардіальну недостатність серця;
в) позаміокардіальну недостатність.
27.5. Дайте коротку характеристику різних патогенетичних варіантів недостатності серця.
I. Недостатність серця від перевантаження розвивається в результаті дії на здорове серце великих навантажень опором або об'ємом, тобто коли збільшується опір серцевому виштовху або приплив крові до певного відділу серця. Це буває при вадах серця, гіпертензії великого або малого кола кровообігу, артеріовенозних фістулах або при виконанні дуже важкої фізичної роботи. При цьому до серця з нормальною скорочувальною здатністю висуваються надмірні вимоги.
II. Міокардіальна недостатність серця розвивається в результаті первинного ураження міокарда. Вона може бути пов'язана з: а) ушкодженням провідникової системи серця (аритмічна) і б) ушкодженням волокон робочого міокарда (міокардіопатична). Причиною її розвитку часто є інфекції, інтоксикація, гіпоксія,
авітамінози, порушення вінцевого кровообігу, деякі спадкові дефекти обміну речовин. При цьому недостатність розвивається навіть при нормальному або зниженому навантаженні на серце.
III. Позаміокардіальна недостатність серця розвивається в результаті дії причин, не пов'язаних безпосередньо з міокардом. її виникнення можуть обумовлювати зменшення припливу крові до серця (гіповолемія, колапс) або перешкоди здійсненню діастоли, у результаті чого серце не може прийняти всю кров, що притікає до нього (накопичення ексудату або транссудату в порожнині перикарда, гостра тампонада серця).
27.6.Які ТИПИ перевантажень серця можуть бути причиною розвитку його недостатності?
Виділяють два типи перевантажень серця.
1. Перевантаження об'ємом виникає тоді, коли до серця або до окремих його порожнин притікає збільшений об'єм крові. У цих умовах серце або його відділ, що зазнає перевантаження, мають переміщати збільшений об'єм крові в артеріальну систему. Це досягається збільшенням хвилинного об'єму серця відповідно до збільшеного венозного повернення.
Перевантаження об'ємом виникає при:
а) збільшенні венозного повернення крові до серця, зокрема при збільшенні об'єму циркулюючої крові (гіперволемія) або збільшенні тонусу венозних судин (зменшення ємності венозної системи);
б) вадах серця — недостатності його клапанів. Так, при недостатності аортального і двостулкового клапанів розвивається перевантаження лівого шлуночка, при недостатності клапана легеневої артерії і тристулкового клапана - перевантаження правого шлуночка.
2. Перевантаження опором виникає тоді, коли серце або окремі його відділи змушені виконувати роботу проти збільшеного опору, що перешкоджає переміщенню всієї крові в артеріальну систему. При перевантаженні опором серце має зберегти свій хвилинний об'єм, незважаючи на збільшений опір вигнанню крові.
Перевантаження опором розвивається при:
а) збільшенні артеріального тиску (збільшенні периферичного судинного опору). При гіпертензії великого кола кровообігу перевантаження опором діє на лівий шлуночок, а при гіпертензії малого кола - на правий шлуночок;
б) вадах серця — стенозах клапанних отворів. 'Так, при стенозі отвору аорти розвивається перевантаження лівого шлуночка, при стенозі отвору двостулкового клапана - лівого передсердя, при стенозі отвору легеневої артерії - правого шлуночка, при стенозі отвору тристулкового клапана - правого передсердя.
27.7.Які механізми можуть забезпечувати компенсацію серця при дії на нього збільшених навантажень?
При дії на серце навантажень об'ємом і опором збільшення роботи серця забезпечується двома типами компенсаторних механізмів.
I. Негайні механізми компенсації серця. До них відносять:
а) гетерюметричтш механізм; б) гомеометричний механізм; в) хроноінотропний механізм;. г) інотропна дія катехоламінів.
II. Механізми довгострокової адаптації серця - гіпертрофія міокарда.