

Practical Plastic Surgery
.pdf332 |
Practical Plastic Surgery |
Maxillary Abnormalities
The most common problems associated with the maxilla include maxillary sagittal hypo/hyperplasia, maxillary vertical hypo/hyperplasia and maxillary transverse hypo/hyperplasia. When evaluating a patient for maxillary surgery the surgeon must evaluate the following:
1.Vertical position of the maxilla
2.Sagittal position of the maxilla
3.Transverse width of the maxilla
Useful measurements include: PNS-Sella, maxillary tooth show, occlusal plane
angle, J point analysis, nasolabial angle, SNA and ANB. The LeFort I osteotomy is used to correct deformities of the maxilla in the vertical, sagittal and transverse planes. It is commonly employed to treat sagittal hypoplasia (small SNA), vertical maxillary excess (excess gingival show) and anterior open bite (large PNS-Sella).
Mandibular Abnormalities
Common problems associated with the mandible include mandibular sagittal hypoplasia and mandibular sagittal hyperplasia. Cephalometric measurements used to evaluate the mandibular position include SNB, ANB, gonial angle and mandibular length. SNB determines the relative position of the mandible to the cranial base, and ANB illustrates the position of the mandible as related to the maxilla. Large values of mandibular length are associated with mandibular prognathism. The patient with a long lower face height or open bite will often have a large gonial angle. The Class III patient often has a diagnosis of mandibular sagittal hyperplasia. Most often, the patient has a prominent lower jaw and chin and a long lower face height. The Class II patient typically has a diagnosis of mandibular sagittal hypoplasia. Most often, the patient has a small mandible, retrusive chin and obtuse
54cervicomandibular angle. Surgical treatment of the mandible for mandibular prognathism includes bilateral sagittal split osteotomy (BSSO) and vertical ramus osteotomy (VRO).
Cephalometric Analysis and Predictions
Computerized cephalometric analysis and predictions have become the standard in orthognathic surgery. Many popular software packages allow for incorporation and analysis of radiographic images. They enable the clinician to pinpoint angles on a digitized AP and lateral cephalogram and generate cephalometric calculations. These calculations can be adjusted for gender, race and age. Following cephalometric analysis, the software generates a cephalometric tracing based on the points and angles identified by the physician. The clinical photograph is overlaid and incorporated into the digitized radiograph. The digitized cephalometric data is used to generate diagnosis and treatment based on the measurements. Overall, the computerized cephalometric software program is a good tool in the rapid diagnosis of dentofacial deformity. It allows for the interface between digitized radiographs, cephalometric measurements and cephalometric predictions. Ultimately, the goal in cephalometric imaging is the incorporation of 3-D imaging and predictions.
Model Surgery
As discussed previously, othognathic surgery consists of mandibular surgery, maxillary surgery and double jaw procedures. Surgeons use model surgery to aid in

Cephalometrics |
333 |
planning of the operation. Model surgery facilitates precise surgical movements. In general, the patient presents for diagnostic evaluation when the skeletal malformation is first identified. At that time, diagnostic records, radiographs, images and dental models are fabricated. This process is repeated immediately before the operation. Appropriate images, measurements, radiographs and models are generated prior to the operation. Specifically, dental casts are fabricated from dental impression and stone. A face-bow record of the patients Frankfort horizontal plane to occlusal plane is generated. The dental casts are articulated using a bite record in centric relation and these are mounted on an anatomic semiadjustable articulator. Reproducible vertical reference lines are placed at the midline, canine tip, mesiobuccal cusp of the first molar and posterior retromolar region. Horizontal reference lines are placed at 10 and 20 mm from the articulator mounting plate. The cast is separated from the mounting ring at 15 mm, centered between the two horizontal reference lines.
Next, model surgery is performed on the mandible, maxilla, or both by positioning the maxillary and mandibular arches into an ideal occlusal relationship. The horizontal and vertical reference lines keep the models in the appropriate vertical, anteroposterior and transverse relationship. A final surgical splint is fabricated using methyl methylacrylate resin. In the case of double jaw surgery, the maxilla is first positioned in the predicted horizontal and vertical position. A splint is fabricated using methylmethacrylate that preserves this “intermediate” maxillomandibular relationship. Next, the mandibular cast is moved into an ideal occlusal relationship with the maxilla, creating the final splint.
Pearls and Pitfalls
An inexperienced surgeon who takes a bite registration in centric occlusion rather than centric relation will end up with inaccurately mounted modes, inaccurate splints, and eventually a nonreproducible, unstable result with a malocclu- 54 sion intraoperatively.
The SN-FH correction must be taken into consideration in order to have an accurate cephalometric prediction and treatment plan. Most patients have a normal SN-FH and thus do not require any correction factors; however children with craniofacial anomalies often require close analysis of the corrected FH, otherwise the cephalometric analysis will not be accurate.
Suggested Reading
1.Ash M. Wheelers Dental Anatomy, Physiology and Occlusion. Philadelphia: Saunders, 2000.
2.Betts N, Turvey T. Orthognathic surgery. In: Fonseca R, ed. Oral and Maxillofacial Surgery. Philadelphia: Saunders, 2000.
3.Ferraro JW. Cephalometry and cephalometric analysis. In: Ferraro JW, ed. Fundamentals of Maxillofacial Surgery. New York: Springer, 1997.
4.Kaban LB, Troulis MJ. Pediatric Oral and Maxillofacial Surgery. Philadelphia: Saunders, 2004.
5.Peterson LJ, Ellis E, Hupp JR et al. Contemporary oral and maxillofacial surgery. St. Louis: Mosby, 1998.
6.Profitt WR, Thomas PM, Camilla-Tulloch JF. Contemporary orthodontics. St Louis, Mosby: 1986.
7.Proffit WR, White RP, Sarver DM. Contemporary treatment of dentofacial deformity. St. Louis. Mosby: 2002.

Chapter 55
Craniofacial Syndromes and Craniosynostosis
Zol B. Kryger and Pravin K. Patel
Craniosynostosis
Introduction
Craniosynostosis is the premature fusion of the sutures of the skull. Although the cause of this condition is not known, TGF-beta has been strongly implicated as playing a major role. Craniosynostosis can occur as an isolated event or in the context of a craniofacial syndrome. The sporadic nonsyndromic cases are more common (incidence of 1 in 2000 live births) than the syndromic cases, many of which are related to defects in the FGF receptor (FGFR). Virchow’s law can help predict the developing skull shape. It states that growth is restricted perpendicular to the fused suture, and compensatory growth occurs parallel to the affected sutures.
Affected Suture
One or more sutures can be affected in craniosynostosis. Sagittal synostosis is the most common form, accounting for over half of all cases. Table 55.1 summarizes the various involved sutures and the characteristic appearance of the skull:
Deformational (Nonsynostotic) Plagiocephaly
This condition is more common than posterior plagiocephaly due to craniosynostosis with an incidence of 1 in 300 live births. The incidence of this condition increased significantly after recommendations by pediatricians that infants sleep supine in order to decrease the risk of SIDS. Deformational plagiocephaly occurs as a result of supine positioning during the first few weeks of life. Features that help distinguish it from synostotic plagiocephaly (lambdoid synostosis) can be seen in Table 55.2.
Table 55.1. Various involved sutures and characteristic appearances of the skull
Involved Suture |
Skull Appearance |
Unicoronal |
Anterior plagiocephaly |
Bicoronal |
Turribrachycephaly (bitemporal widening) |
Sagittal |
Scaphocephaly (biparietal narrowing) |
Metopic |
Trigonocephaly (triangular forehead) |
Lambdoid |
Posterior plagiocephaly (flat posterior skull) |
Coronal, lambdoid, metopic |
Kleeblattschadel (clover leaf skull) |
Practical Plastic Surgery, edited by Zol B. Kryger and Mark Sisco. ©2007 Landes Bioscience.

Craniofacial Syndromes and Craniosynostosis |
335 |
|
|
|
|
Table 55.2. Comparison of features between synostotic
|
and deformational plagiocephaly |
|
|
|
|
|
Synostotic |
Deformational |
Feature |
Plagiocephaly |
Plagiocephaly |
Sutures |
Fused |
Patent |
Eyebrow |
Elevated on affected side |
Eyebrow lower on affected side |
Ear |
Rotated anterosuperior |
Rotated posteroinferior |
Nose |
Deviated to opposite side |
Deviated to affected side |
Chin |
Deviated to opposite side |
Deviated to affected side |
Cheek |
Forward on affected side |
Flattened on affected side |
Torticollis |
Contralateral side |
Ipsilateral side |
Associated Symptoms |
|
Elevated intracranial pressure (ICP)—occurs in 13% of single suture synostosis |
|
and 42 % of multiple suture synostoses. |
|
Strabismus—seen most often in unilateral, coronal synostosis. It is due to paresis |
|
of the superior oblique muscle. |
|
Torticollis—seen in about 15% of cases of anterior plagiocephaly, usually on the |
|
unaffected side. |
|
Cognitive deficits—seen most commonly with metopic synostosis. |
|
Treatment |
|
The goal of treatment is foremost to allow adequate space for the brain to grow. |
|
Of lesser importance is the creation of an aesthetically normal skull and forehead. |
|
Molding (orthotic cranioplasty) |
55 |
Molding of the skull, or orthotic cranioplasty, is done with a helmet worn up to 23 hours a day for 2-4 months. It should begin around the age of 6 months and always before the age of 14 months. It is the recommended treatment for deformational plagiocephaly and can also be used as an adjunct to surgery.
Surgery
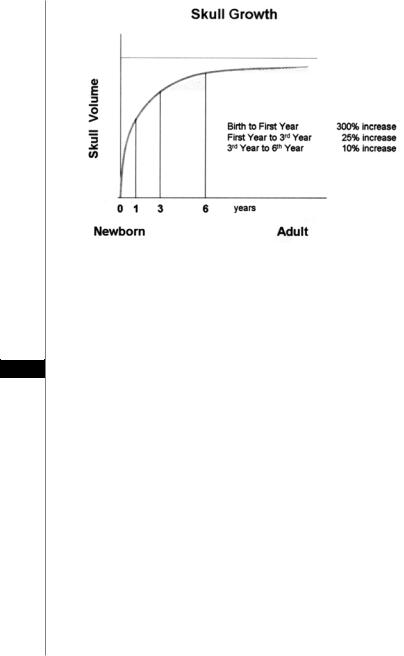
Since the most rapid phase of skull growth occurs within the first year of life (Fig 55.1), early treatment is required. Surgical intervention is most commonly performed between the ages of 3 to 12 months of age. There is some evidence that earlier intervention results in fewer learning disabilities and emotional problems in later years. However, these findings are disputed by those who believe that operating on infants with craniosynostosis will not have an effect on their future cognitive abilities. Regardless of the timing, surgical treatment must be tailored to the individual’s deformed structures.
Unilateral coronal synostosis is managed with unilateral or bilateral fronto-or- bital advancement. Bilateral coronal synostosis is managed with bilateral frontoorbital advancement. If there is evidence of brachycephaly, total calvarial reconstruction may be required. Sagittal synostosis is treated with strip craniectomies and partial-wedge osteotomies. If biparietal expansion is required, anterior and posterior parietal wedges may be required. Lambdoid synostosis is managed with excision of the lambdoid suture. Metopic synsotosis is treated with removal of the supraorbital bar, corticotomy and correction of the midline angle with bone grafts
336 |
Practical Plastic Surgery |
|
|
|
|
|
|
|
Figure 55.1. The growth curve of the skull.
or miniplates. Kleeblattschadel is treated after life-threatening conditions are addressed (e.g., hydrocephalus, airway obstruction) with anterior calveriectomy and fronto-orbital advancement. Excess constricting bands of bone are removed.
Complications
As might be expected from the magnitude of the procedures, the complications
55can be devastating. Early complications specific to these procedures include: sagittal sinus tears with venous infarction, subdural hematoma, cerebral edema, excess vasopressin production (SIADH), nerve injuries, injury to the orbits, infections, elevated ICP and dural leaks. Late complications include: incomplete advancement, palpable hardware, alopecia, asymmetry, orbital deformities, pseudomeningocele and increased ICP.
Craniofacial Syndromes
Many craniofacial syndromes have been described in the literature. However, with a few exceptions, these are rare syndromes seen primarily in specialized centers. A discussion of the more commonly encountered craniofacial syndromes follows with an emphasis on the similarities and differences between them.
Apert Syndrome (Acrocephalosyndactyly)
•Autosomal dominant
•Bicoronal synostosis
•Hypertelorism or exorbitism
•Strabismus
•Maxillary hypoplasia
•Acne and coarse skin

Craniofacial Syndromes and Craniosynostosis |
337 |
•Large ear lobes
•Syndactyly of all fingers (mitten hands) and toes
•Variable mental retardation
Crouzon Syndrome (Craniofacial dysostosis) |
|
||
• Autosomal dominant |
|
||
• |
Bicoronal (occasionally sagittal) synostosis |
|
|
• |
Hypertelorism or exorbitism |
|
|
• |
Maxillary hypoplasia |
|
|
• |
Micrognathia |
|
|
• |
No abnormalities of the hands or feets |
|
|
• |
Normal intelligence |
|
|
Pfeiffer Syndrome (Acrocephalosyndactyly type V) |
|
||
• Autosomal dominant |
|
||
• |
Craniosynostosis of multiple sutures |
|
|
• |
Maxillary hypoplasia |
|
|
• |
Broad thumbs and great toes |
|
|
• |
Brachydactyly with partial syndactyly |
|
|
• |
Normal intelligence |
|
|
Saethre Chotzen Syndrome (Craniocephalosyndactyly) |
|
||
• Autosomal dominant |
|
||
• |
Bicoronal synostosis |
|
|
• |
Maxillary hypoplasia |
|
|
• |
Strabismus |
|
|
• |
Vertebral anomalies |
|
|
• |
Low hairline |
|
|
55 |
|||
• |
Palatal abnormalities (often a cleft) |
||
•Brachydactyly with syndactyly
•Normal intelligence
Carpenter Syndrome
•Autosomal recessive
•Craniosynostosis
•Deafness
•Brachydactyly with syndactyly
•Polydactyly (preaxial)
•Mental retardation
Treacher-Collins Syndrome (Mandibulofacial dysostosis)
•Autosomal dominant
•Prominent eyelid abnormalities (e.g., pseudocoloboma and absence of lashes)
•Downward sloping palpebral fissures
•Poorly developed orbital rims
•Midface (zygomaticomaxillary) hypoplasia
•Macrostomia and resulting airway distress
•Cleft palate

338 |
Practical Plastic Surgery |
Velocardiofacial Syndrome
•Autosomal dominant
•Cleft palate or submucus cleft
•Cardiac abnormalities
•Ectopic carotid arteries
•Malar flattening
•Prominent broad nose
•Epicanthal folds
•Vertical maxillary excess
•Retrognathia
•Developmentally delayed
Pierre Robin Sequence
•Retrognathia (occasionally described as retromicrogenia)
•Glossoptosis
•Airway obstruction
•Variable cleft palate
Although not a true craniofacial syndrome, Pierre Robin sequence is an impor-
tant constellation of findings. This condition can be life-threatening due to airway compromise. Initial treatment consists of prone positioning, while surgical management is reserved for more severe cases.
Pearls and Pitfalls
•Treatment of craniosynostosis and craniofacial syndromes requires a multidisciplinary team approach. Early involvement of the other relevant specialties (neurosurgery, otolaryngology, oral surgery, speech therapy, a prosthodon-
tist and a geneticist) is essential for achieving a good outcome.
55• Prevention of deformational plagiocephaly is quite simple. It consists of turning the infant’s head from side to side several times throughout the day, and placing the infant prone while he is awake and under observation by the caretaker.
•In any infant suspected of having a craniofacial abnormality, evaluation of the airway is the most important initial step in the work-up.
•Distraction osteogenesis of the facial skeleton is an alternative to bone grafting that offers excellent long-term outcomes. It should be considered whenever greater than 10-15 mm of advancement of the facial bones is required.
•Crouzon’s syndrome can be distinguished from Apert and Pfeiffer syndromes by the absence of hand abnormalities.
•Almost all of the craniofacial syndromes are transmitted via an autosomal dominant pattern with variable expression. An important exception is Carpenter syndrome.
•The FGFR is known to be mutated in the majority of the craniofacial syndromes involving craniosynostosis. Future therapeutic approaches may target these mutations.

Craniofacial Syndromes and Craniosynostosis |
339 |
Suggested Reading
1.Anderson FM, Geiger L. Craniosynostosis: A Survey of 204 cases. J Neurosurg 1965; 22:229.
2.Breugem CC, van R, Zeeman BJ. Retrospective study of nonsyndromic craniosynostosis treated over a 10-year perior. J Craniofac Surg 1999; 10:140.
3.Tessier P. The definitive plastic surgical treatment of the severe facial deformities of craniofacial dysostosis. Crouzon’s and Apert’s diseases. Plast Reconstr Surg 1971; 48:419.
4.Huang MH, Gruss JS, Clarren SK et al. The differential diagnosis of posterior plagiocephaly: True lambdoid synostosis versus positional molding. Plast Reconstr Surg 1996; 98:765.
5.Kapp-Simon KA. Mental development and learning disorders in children with single suture craniosynostosis. Cleft Palate Craniofac J 1998; 35:197.
6.Losken HW, Pollack IF. Craniosynostosis. In: Bentz ML, ed. Pediatric Plastic Surgery. 1st ed. Stamford: Appleton and Lange, 1998.
7.Marchac D, Renier D, Broumand S. Timing of treatment for craniosynostosis and faciocraniosynostosis: A 20-year experience. Br J Plast Surg 1994; 47:211.
8.Mulliken JB, Vander Woude Dl, Hansen M et al. Analysis of posterior plagiocephaly: Deformational versus synostotic. Plast Reconstr Surg 1999; 103:371.
9.Noetzel MJ et al. Hydrocephalus and mental retardation in craniosynostosis. J Pediatr 1985; 107:885.
10.Persing JA, Jane JA, Edgerton MT. Surgical treatment of craniosynostosis. In: Persing JA, Edgerton MT, Jane JA, eds. Scientific Foundation of Surgical Treatment of Craniosynostosis. Baltimore: Williams and Wilkins, 1989.
55

Chapter 56
Craniofacial Microsomia
Zol B. Kryger
Introduction
Craniofacial microsomia, also termed hemifacial microsomia, is defined as a congenital hypoplasia of the facial skeleton and soft tissues. There is no known genetic cause. It is thought to be due to an in utero vascular insult in the developing first or second branchial arches. It is unilateral in 90% of cases. After cleft lip and palate, this condition is one of the more common craniofacial congenital abnormalities. The incidence in the U.S. is estimated to be 1 in 5,000 births.
Clinical Findings
Any of the skeletal, nervous, and soft tissue structures derived from the first and second branchial arches can be affected. Consequently, there is a wide spectrum of presenting signs and symptoms.
Microtia and inner ear abnormalities are usually present on examination. One may notice abnormalities of the auricle with preauricular skin tags on the affected side. Examination of the lower face reveals a cant of the occlusion plane sloping downward and away from the hypoplastic side. The chin is deviated towards the affected side, and the distance from the oral commissure to the ear may be shortened.
A full examination of oral cavity, including the teeth and dental occlusion, should be performed. The muscles of mastication are often dysfunctional; however, their involvement is not always proportional to the degree of mandibular hypoplasia. This dysfunction may be manifested in difficulty opening the mouth. The lateral pterygoid is also usually involved, causing an inability to deviate the chin to the contralateral side. Soft tissue and palatal clefts may also be present.
A complete cranial nerve examination is essential since patients may also demonstrate neurologic abnormalities. The marginal mandibular branch of the facial nerve is most commonly involved. Cerebral abnormalities are rare.
Radiographic evaluation has traditionally included cephalograms and a panorex. Computed tomography with 3-D reconstructions will give the most precise information to evaluate the skeletal abnormalities and assist in the preoperative planning.
Skeletal Abnormalities
The mandible is most commonly involved in craniofacial microsomia. The degree of mandibular hypoplasia has been classified by Pruzansky:
•Type I. Mild hypoplasia of the condyle and ramus; the body is unaffected. The TMJ is functional. There are minimally noticeable morphological changes.
•Type IIA. The condyle and ramus are severely hypoplastic. The coranoid process can be absent. The condyle maintains a normal position relative to the glenoid fossa.
Practical Plastic Surgery, edited by Zol B. Kryger and Mark Sisco. ©2007 Landes Bioscience.

Craniofacial Microsomia |
341 |
•Type IIB. Similar to Type IIA, yet the condyle and glenoid fossa are not in the normal position and plane.
•Type III. The condyle and ramus are absent and there is no TMJ.
Other bones of the craniofacial skeleton can be hypoplastic as well. The maxilla
is reduced in the vertical plane, and contributes to the cant of the occlusion plane. The zygoma and zygomatic arch are often decreased in length and the arch may be entirely absent. The temporal and frontal bones are affected less; however the orbits are often reduced in all dimensions leading to microphthalmos.
Classification of Unilateral Craniofacial Microsomia
There are a number of classification schemes. The two that are common and easy to use are the OMENS classification (Table 56.1), and the system proposed by Munro and Lauritzen in 1985 (Table 56.2).
Treatment by Age
There is no uniform treatment plan that is appropriate for everyone. Children under the age of two with craniofacial microsomia do not usually undergo major reconstructive surgery. Preauricular skin tags or other cartilaginous remnants may be excised. Commissuroplasty can correct those patients with macrostomia at this young age.
After the age of two, distraction osteogenesis can correct mandibular ramus hypoplasia as seen in Pruzansky Type I and IIA. This will both lengthen the mandible and improve the function of the muscles of mastication. Bilateral cases can be treated with bilateral distraction, allowing closure of tracheostomies in many children.
At the age of four, children with Pruzansky Type III mandibular hypoplasia undergo costochondral rib-graft reconstruction of the mandible combined with a LeFort I osteotomy and sagittal split of the contralateral mandibular ramus. The zygomatic arch and glenoid fossa are also reconstructed using a rib graft and a cap of costo-
chondral cartilage, respectively. 56 After the age of six, and microtia repair may be performed and orthodontic
treatment initiated. Augmentation of the facial soft tissue with muscle flap transfers is also occasionally necessary at this age. Once skeletal maturity has been reached in the early teenage years, residual deficiencies should be addressed.
Teenagers who have not undergone any prior treatment will require alternative types of reconstruction. Interpositional bone grafts can be used to correct mild mandibular length deficiencies. More severe microsomia can be treated with combined LeFort I osteotomy, bilateral sagittal split of the mandible, and genioplasty. Micrognathia can be treated with bilateral mandibular advancement.
Table 56.1. The “OMENS” classification for craniofacial microsomia
|
Involved Structure |
Description |
O |
Orbit |
Size and position of the orbit |
M |
Mandible |
Degree of mandibular hypoplasia |
E |
Ear |
Extent of microtia |
N |
Facial nerve |
Which branches are involved |
S |
Soft tissue |
Degree of muscular and subcutaneous deficiency |
|
|
|