

5. The chemistry of ionized, protonated and cationated amines in the gas phase 225
atoms of the ˛-methylene group were always retained in the ionic product. Thus, (CH3)3CCD2NH2Cž loses C4H7ž [presumably the 2-methyallyl radical], with formation of CHD2NH3C . Support for this mechanism was adduced from the effects of progressive methylation on nitrogen on the relative abundance of ions formed by DHT and ˛-cleavage of ionized neopentylamines, the former process being sufficiently favoured energetically to dominate the latter only in the case of (CH3)3CCH2NH2Cž . Moreover, a similar energetic analysis of ionized isobutyl alcohol, the derived methyl ether and the analogous amine had previously concluded that DHT is circumvented for (CH3)2CHNH2Cž as the corresponding INC, [(CH3)2CHž CH2DNH2C ] is inaccessible at low internal energies because it was insufficiently stabilized relative to the separated products116.
However, a later analysis115 disputed this conclusion and proposed instead that DHT in ionized neopentylamine proceeds via the DI, (CH3)2(CH2ž )CCH2NH3C , which then undergoes bond fission to the INC, [(CH3)2CDCH2 ž CH2NH3C ], containing an ˛-DI. Subsequent hydrogen transfer then leads to CH2D(CH3)C CH2ž and CH3NH3C , possibly via the INC, [CH2D(CH3)C CH2ž CH3NH3C ] (Scheme 12). This mechanism also accounts for the occurrence of C2H5ž loss from metastable (CH3)2CHCH2NH2Cž , which occurs after skeletal rearrangement of the -DI CH3(CH2ž )CHCH2NH3C (Scheme 13).
H
+ |
1,4-H |
+ |
|
|
+ |
|||
NH2 |
NH3 |
|
|
|
H2C |
|
NH3 |
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
+
+ H3C NH3
SCHEME 12
This second alternative is more consistent with the general behaviour of ionized alkylamines, many of which isomerize rapidly to DIs. Thus, extensive exchange of the hydrogen atoms attached to nitrogen with those of the methyl groups precedes butenyl radical loss from ionized labelled neopentylamines116.
Most hydrogen transfers and other isomerizations of ionized alkylamines generally occur via DIs rather than INCs82. However, the participation of an INC in DHT remains an attractive proposition, even if it occurs after the first hydrogen transfer has taken place to give a DI. DHT involving an intact N-alkyl chain and eventual C N cleavage may also occur via isomerization to a ˇ-DI, with production of an ammonium (or alkylammonium) cation42.
As with pseudo-˛-cleavage, the suppression of SHT and DHT by N-alkylation reflects the reduced tendency of ionized secondary and especially tertiary amines to undergo the initial hydrogen transfer steps to form DIs44. This common trend also supports the contention that ionized amines generally isomerize via DIs.
c. Other reactions. Certain routes for alkyl radical expulsion which are not ˛-cleavages cannot readily be interpreted as pseudo-˛-cleavages. Thus, ionized 1-butylamine loses

226 |
|
|
|
|
Richard D. Bowen |
|
|
|
|
+ |
1,4-H |
+ |
|
+ |
|||||
|
NH2 |
NH3 |
|
|
|
NH3 |
|||
|
|
|
|
||||||
H |
|
|
1,2-CH2 NH3 |
+ |
|
|
|||
|
|
|
|
||||||
|
|
|
|
|
|
|
|||
+ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
NH2 |
|
|
+ |
|
+ |
|||
|
|
|
|
|
|
||||
|
|
steps |
NH3 |
||||||
|
|
|
|
NH3 |
|||||
|
|
|
|
|
|
|
|
|
|
|
α |
|
|
|
1,4-H |
||||
|
|
|
|
||||||
|
|
|
|
|
+ |
||||
|
|
|
|
|
|
|
|||
+ |
|
|
|
|
|
|
|
NH2 |
|
|
NH2 + |
C2H5 |
|
|
|
|
|
||
H
SCHEME 13
C2H5ž . The chain length is insufficient to permit the hydrogen transfers needed for pseudo- ˛-cleavage. Low-energy multiphoton dissociation ionization studies117 show that ionized 1-butylamine has predominantly isomerized via a υ-DI to ionized 2-butylamine, which may then fragment via ˛-cleavage. One route for this rearrangement involves complexes containing C4H8Cž species (Scheme 14). A mechanistically attractive alternative is isomerization of the appropriate -DI via 1,2-CH2 NH3C shifts (see Schemes 7 and 13).
H |
+ |
+ |
|
|
|
||
|
NH2 1,5-H |
NH3 |
NH3 |
|
|
+ |
+ |
1,4-H |
+ |
+ |
|
+ NH3 |
|
NH3 |
|
|||
NH2 |
NH3 |
|
|
|
|
|
|
SCHEME 14 |
|
|
|
4. Reactions of immonium ions derived from ionized alkylamines
The reactions of CnH2nC2NC immonium ions generated by dissociative ionization of alkylamines have been reviewed recently118, so only a brief summary is presented. The principal reaction is loss of an alkene, Cm H2m , with formation of a smaller immonium ion3,68. Some lower members of the homologous series (n D 2 5) lose an alkyne, Cm H2m 2, to give a protonated alkylamine119 121, but this fragmentation is rapidly superseded by alkene elimination for larger CnH2nC2NC species.

5. The chemistry of ionized, protonated and cationated amines in the gas phase 227
Loss of molecular hydrogen is also important for certain immonium ions with at least one N-methyl group and a hydrocarbon chain containing one hydrogen atom less than a complete alkyl group attached to the nitrogen atom. The archetypal example is CH3CHDNHC CH3. This reaction, which occurs both in fast fragmentations in the ion source and in the dissociation of metastable ions, is found by 2H-labelling to occur with a 1,4-regioselectivity. Thus, CH3CHDNDC CH3 loses H2, but CH3CHDNHCCD3 eliminates HD; CD3CHDNHCCH3 loses both H2 and HD, after exchange of the protium and deuterium atoms in the C2HD3 entity has taken place122. This unusual fragmentation, which is rationalized by equation 11, occurs with a large and relatively specific kinetic energy release. Furthermore, the overall isotope effects on the rate of H2 and HD loss from labelled analogues in which either N CD2 D or C CD2 D bonds must be broken are similar (ca 1.7:1)122, thus suggesting that these two C H bonds are broken in concert in the transition state118. Both the regioselectivity and the association of a large KE release with a 1,4-elimination are extremely unusual, if not unique, for loss of molecular hydrogen from isolated organic ions. Part of the reason for this behaviour may lie in the stability of the resultant unsaturated immonium ion (e.g. CH2DCHNC HDCH2, formed by 1,4-H2 loss from CH3CHDNHCCH3). In contrast, the smallest immonium ion, CH2DNH2C , is distinct among immonium ions in losing molecular hydrogen with a large KE release via a route involving 1,2-elimination123. It is this route, rather than 1,4-elimination, which is usual for most small organic ions. In any event, the observation of significant CnH2nNC ions is often analytically useful because H2 elimination is associated with the RCH2CHDNHCCH3, CH3C(R)DNHCCH3, RCH2CHDNC (CH3)2, CH3C(R)DNHC(CH3)2 and similar structures.
R |
|
|
|
R |
|
|
|
|
H |
|
|
+ H2 |
(11) |
|
N |
H |
|
|
||
|
|
|
||||
|
|
|
N |
|
||
R |
+ |
|
|
R |
+ |
|
There are at least six different routes for alkene elimination from CnH2nC2NC species. However, the two most important involve loss of an alkene derived from an intact N-alkyl substituent.
The first route results in expulsion of Cm H2m with associated hydrogen transfer from the N R group (R D CnH2mC1) to nitrogen. Typically, the alkene is derived from the largest alkyl substituent. Thus, for example, CH2DNC (Cm H2mC1)2 eliminates Cm H2m with formation of CH2DNHCCm H2mC1; similarly, CH2DNHC Cm H2mC1 loses Cm H2m to produce CH2DNH2C . This fragmentation has traditionally been rationalized as a four-centre reaction (equation 12) despite the fact that it has been known for many years that specific ˇ-hydrogen transfer does not always occur, in either fast124 or slow125 dissociations of this general class126.
R |
+ |
H2 |
|
R |
+ |
|
|
|
C |
|
|
|
|
||||
|
|
|
|
|
(12) |
|||
|
N |
CHR |
|
|
NH2 + H2C |
|
CHR |
|
|
|
|
|
|||||
|
|
|
|
|||||
R |
H |
H |
|
R |
|
|
|
|
|
|
|
|
|
|
|
More recent work93,127 129 has shown that ˇ-hydrogen transfer takes place only if the cation corresponding to the N R substituent is stable. Thus, CH2DNC (CH3)CH(CD3)2 and related species expel C3HD5, via specific D-transfer128,129, because isopropyl cation is stable with respect to a 1,2-H shift. In contrast, if the associated cation readily rearranges to a more stable isomer, there is actually a discrimination against

228 Richard D. Bowen
ˇ-hydrogen transfer, as is shown by the loss of predominantly (79%) C3H4D2 from CH2DNC(CH3)CH2CD2CH393,128,129, which would be expected to expel only C3H5D via the traditional mechanism. This contrasting regioselectivity is intelligible if the fragmentation is INC-mediated118,127 129. Furthermore, this mechanism also explains why the dissociation of immonium ions by this route is accompanied by only a moderate KE release (T1/2, the KE release estimated from the width at half-height of the corresponding
Gaussian metastable peak, is typically ca 2 3 kJ mol 1).
When stretching, the N C bond leads to a stable carbocation, a unidirectional hydrogen transfer to nitrogen occurs from one of the original ˇ-carbon atoms, without rearrangement of the developing cation. Consequently, the loss of C3HD5
from CH2DNC (CH3)CH(CD3)2 |
and |
homologous |
immonium |
ions is explained |
||||||||||||||
(equation 13)128,129. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
+ |
|
|
|
|
|
H |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
+ |
|
|
|
||||||
|
|
|
|
N |
+ |
|
|
|
|
|
|
|
|
|
|
|||
N |
|
|
|
|
|
|
|
|
|
|
|
NH |
(13) |
|||||
|
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
+ |
+ |
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
NH |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
On the other hand, when the initial bond elongation would lead to an unstable carbocation such as a primary carbonium ion, isomerization of the developing cation occurs, via a 1,2-H shift, with formation of an INC containing a rearranged carbocation. The positive charge in this isomerized cation generally is located on the carbon atom that was in the ˇ-position in the initial N R group. The subsequent hydrogen transfer to nitrogen occurs from a carbon atom next to that carrying the formal cationic site. These carbon atoms are those which were in the ˛- and -position in the original N R substituent. Therefore, the strong preference for apparent ˛- and-hydrogen transfer in CH2DNC (CH3)CD2CH2CH3, CH2DNC(CH3)CH2CD2CH3 and CH2DNC(CH3)CH2CH2CD3 is explained128,129. The minor residual contribution from apparent ˇ-hydrogen transfer reflects the migration of one hydrogen atom from the ˇ- to the ˛-carbon during the isomerization step. Thus, CH2DNC (CH3)CH2CD2CH3 rearranges to CH2DNC (CH3)CH(CH2D)CH3, which undergoes H- and D-transfer in the ratio 5:1, to a first approximation (Scheme 15).
Further important aspects of this mechanism include the occurrence of a small isotope effect (ca 1.08:1, overall) favouring H-migration in the developing CH2CH2CH3 entity of CH2DNC(CH2CH2CH3)CH2CD2CH3 over the corresponding D-migration in the putative CH2CD2CH3 entity128,129. This finding indicates that the 1,2-H/D shift is beginning to occur as the appropriate N C bond is stretched on the way to the transition state for the reaction, thus circumventing formation of an unstable n-propyl cation. Exactly parallel effects were found in alkene loss from protonated n-propylamines, thus underlining the generic nature of this type of INC-mediated process130. Moreover, the minor deviation of the ratios of ˛-, ˇ- and -hydrogen transfer from those (2:1:3) expected from the first-order approximation was explained by invoking a small preference factor (ca 1.13:1) favouring transfer from the original ˛-carbon128. This preference factor arises because the isopropyl cation formed by isomerization of the n-propyl entity does not enjoy full rotational freedom; consequently, hydrogen transfer from the methyl group derived from

5. The chemistry of ionized, protonated and cationated amines in the gas phase 229
|
|
D |
D |
+ |
|
|
|
|
|
|
+ |
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
||||
N |
|
|
|
|
|
NH D |
|
|
|
|
|
NH + C3 H4 D2 |
|||
|
|
|
|
|
|
|
|
||||||||
+ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
D |
|
|
|
|||
|
|
|
|
|
|
|
|
2 parts |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
H |
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
D |
D |
|
|
|
|
H |
H |
+ |
|
|
|||
|
|
|
|
|
|
+ |
|
|
|||||||
N |
+ |
|
|
|
N D |
|
3 parts |
|
NH D |
||||||
|
|
|
|
|
|
H |
D |
|
|
D |
|||||
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
H |
|
|
|
|||
|
|
|
|
|
|
|
|
1 part |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
+ |
|
|
|
+ |
|
|
|
|
|
|
+ |
|
|
||
|
|
|
|
|
|
|
|
|
+ C3 H4 D2 |
||||||
NH |
|
+ C3 H5D |
|
|
ND D |
|
|
|
|
NH |
|||||
|
|
|
|
|
|||||||||||
SCHEME 15
the ˛-methylene group is slightly favoured because the nitrogen atom is initially closer to this methyl group than the more distant -methyl group.
The second general fragmentation of immonium ions results in loss of an alkene containing fewer carbon atoms than the N R substituent. Usually, Cm 1H2m 2 is lost from CH2DNC (Cm H2mC1)2 or CH2DNHC Cm H2mC1. This route occurs less frequently that the alternative involving Cm H2m elimination, but it remains a common reaction3,118. This second fragmentation differs substantially from the first in showing a pronounced regioselectivity (>99%) involving 1,5-hydrogen transfer93,128,129.
Thus, CH2DNC (CH3)CH2CH2CD2CH2CH3 is unique among 2H-labelled analogues of CH2DNC(CH3)CH2CH2CH2CH2CH3 in undergoing C4H7D loss via 1,5-D transfer118. These data are consistent with fragmentation via a pericyclic process isoelectronic with the retro ‘ene’ reaction131.
R3 |
|
R3 |
|
R3 |
R N + |
R N |
+ |
|
R N + + H2C |
|
||||
R2 |
|
R2 |
|
R2 |
H |
|
H |
|
|
(14) Several sources of information indicate that this fragmentation is better described as a two-step process (equation 14), in which the 1,5-H shift is largely or wholly completed before C C cleavage occurs. Thus, both the KE release accompanying this dissociation and the extent to which it competes with the alternative fragmentation in which Cm H2m is lost are strongly influenced by the degree of substitution at the -carbon atom. As this carbon atom becomes more heavily substituted, the KE release decreases sharply and Cm 1H2m 2 elimination competes more effectively with Cm H2m loss127. These trends are
230 |
Richard D. Bowen |
sensible if the mechanism involves a substantial build-up of positive charge on the - carbon atom during the course of the reaction. Furthermore, competition experiments in alkene elimination from CH2DNC R1R2 ions, in which the 1,5-H shift may occur from a -carbon atom in either of the two N-alkyl substituents, show that the hydrogen is transferred essentially exclusively (>99% selectivity) from the more heavily substituted site132. Thus, CH2DNC(CH2CH2CH3)CH2CH2CH2CH3 expels C3H6 with a large KE release (T1/2 D 48 kJ mol 1) from the n-butyl group, rather than C2H4 (which would be expected to have a very large KE release) from the n-propyl substituent (Scheme 16).
|
|
+ |
|
|
|
N |
N |
N |
|
+ |
CH2 |
CH2 |
CH2 |
+ |
|
H |
H |
|
|
CH2 |
H2 C + |
+ |
CH2 |
CH2 |
N |
N |
+ |
||
|
+ |
|
|
|
CH2 |
CH3 |
CH3 |
|
|
SCHEME 16
The large KE releases that typically accompany alkene loss via this channel are further evidence that the reaction involves an intermediate or transition state of substantially higher energy than the combined enthalpy of the products. Loss of C2H4 usually gives rise to a very broad dished metastable peak (T1/2 ca 70 kJ mol 1); elimination of C3H6 normally produces a similar peak when it may be formulated to proceed via a primary cation, but a rather less broad, flat-topped peak (T1/2 ca 50 kJ mol 1) when it can occur via a secondary cation. The peak for expulsion of C4H8 via a tertiary cation remains broad, but generally it is not flat-topped, and the KE release is reduced still further (T1/2 ca 25 kJ mol 1)127,132. Typical examples of the various peak shapes for alkene loss from immonium ions are shown in Figure 3.
In addition to the influence of the level of substitution at the -carbon atom, progressive homologation also diminishes the KE release. Thus, the T1/2 values for Cm 1H2m 2 loss from CH2DNC (CH3)CH2(CH2)m 2CH3 ions decrease sharply from 70 kJ mol 1 for CH2DNC (CH3)CH2CH2CH3 (m D 3, primary cation at the -carbon atom in the
intermediate |
or transition state), to 45 kJ mol 1 for |
CH2 |
D |
NC (CH3)CH2CH2CH2CH3 |
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
127 |
|
|
further homologation |
|
(m D 4, secondary cation at the -carbon atom) |
. However, |
||||||||||||||||
results in a more |
gradual reduction (T1/2 D 33, 19, 11, 9.6, 8.9 |
and 7.0 kJ mol 1 for |
|||||||||||||||
m |
D |
5 |
, |
6 |
, |
8 |
, |
10 |
, |
14 |
and 18, respectively) because the substitution level at the -carbon |
||||||
|
|
|
|
|
|
133 |
. This progressive decline, which is accompanied by a gradual |
||||||||||
atom remains constant |
|
||||||||||||||||
change in peak shape from flat-topped to approximately Gaussian profile, has been interpreted in terms of a ‘degrees of freedom’ effect133. As the eliminated alkene increases in size, there are more ways of distributing energy in the vibrational and rotational modes, so the partitioning of reverse critical energy as translational (i.e. KE release) is diminished. Moreover, the estimated reverse critical energy for the reaction declines from 173 (m D 3) to 70 kJ mol 1 (m D 18) as the homologous series of ions is ascended; consequently, the total amount of energy, some of which may be partitioned as translation, also decreases.

5. The chemistry of ionized, protonated and cationated amines in the gas phase 231
+ |
+ |
[33.6] |
+ |
+ |
[51.8] |
N |
HN |
N |
N |
|
|
m / z |
|
|
|
m / z |
32 |
33 |
34 |
50 |
51 |
52 |
53 |
|
(a) |
|
|
|
(b) |
|
+ |
+ |
[70.4] |
|
+ |
+ |
[76.4] |
N |
N |
|
N |
N |
|
|
m / z |
m / z |
69 |
70 |
74 |
78 |
|
(c) |
|
(d) |
FIGURE 3. Metastable peaks for loss of (a) C3H6 from CH2DNC (CH2CH3)CH2CH2CH3, (b) C2H4 from CH2DNC (CH2CH3)CH2CH2CH3, (c) C3H6 from CH2DNC (CH2CH2CH2CH3)2 and (d) C4H8
from CH2DNC [CH2CH2CH(CH3)2]2. Reproduced by permission of The Royal Society of Chemistry from R. D. Bowen, J. Chem. Soc., Perkin Trans. 2, 409 (1982)
C. Reactions of Ionized Unsaturated and Cyclic Amines
In contrast to ionized alkenols, which have been extensively investigated133, radicalcations derived from alkenylamines have received much less detailed attention. This omission, which partly reflects the relative difficulty in synthesizing unsaturated amines, particularly if incorporation of 2H- or 13C-labels is required, is unfortunate because the chemistry of ionized alkenols134 and the derived ionized ethers, which have been studied more recently135 138, is rich and interesting.
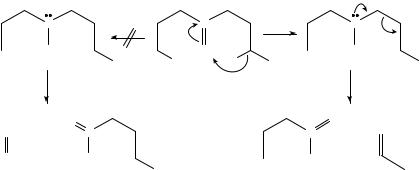
The limited data which are available suggest that ˛-cleavage of the saturated N-alkyl substituent(s) continues to dominate the fragmentations of ionized alkenylamines. Thus, CH2DCHCH2NHCH2CH3 and CH2DCHCH2NHCH2CH2CH3 serve as precursors for the unsaturated immonium ion CH2DCHCH2NHC DCH2, via ionization and elimination of CH3ž and C2H5ž , respectively139. At low internal energies, this immonium ion eliminates HCN, via DHT, and a minor amount of C3H6, via SHT, in sharp contrast to its saturated analogue, CH3CH2CH2NHCDCH2, which loses C2H4 and C3H6. Thus, the presence of the double bond appears to modify the chemistry in the ionized amine (˛-cleavage of the double bond is obviously difficult) and the derived immonium ion (the usual channels for alkene elimination are disrupted because the allyl group has quite different properties from the n-propyl substituent). The loss of C3H6 from both CH3CH2CH2NHC DCH2 and CH2DCHCH2NHCDCH2 is deceptive: these fragmentations are not analogous to one
232 |
Richard D. Bowen |
other. The former entails hydrogen transfer from the largest N-alkyl substituent to the nitrogen atom; the latter involves hydrogen transfer in a different direction, from the isolated methylene group of CH2DCHCH2NHC DCH2 to the terminal methylene group of the allyl substituent. This process presumably proceeds via a 1,5-H shift, but this deduction has not yet been confirmed by labelling experiments. The analogous process to C3H6 expulsion from CH3CH2CH2NHC DCH2 would be C3H4 loss from CH2DCHCH2NHC DCH2; however, this reaction requires formal proton abstraction from an incipient allyl cation, which would give rise to an unfavourable geometry of allene in which the p-orbitals of the developing new -bond are orthogonal140,141.
Many intriguing possibilities exist for isomerization of ionized alkenylamines to DIs. A detailed study of the chemistry of metastable ionized allylamine and cyclopropylamine showed that both these species rearrange readily to the DI žCH2CH2CHDNH2C , which is accessible via a 1,2-H shift or ring opening by C C cleavage, respectively (Scheme 17)142. Previous electron spin resonance (ESR) studies at 77 K reveal that this DI is formed in condensed phases from both ionized allylamine and cyclopropylamine143. In the gas phase, loss of a hydrogen atom is the major fragmentation; this process was shown
by CID experiments to give the conjugated immonium ion, CH2DCHCHDNH2C142 . Thermochemical data were used to construct a potential energy profile (PEP144) for the system, which showed that ionized allylamine and cyclopropylamine had comparable energies, that were estimated to be some 80 kJ mol 1 above that of žCH2CH2CHDNH2C142 . Loss of a hydrogen atom was interpreted as ˇ-cleavage of this DI.
+ |
|
|
|
+ |
|
|
|
+ |
|
NH2 |
α |
|
H + |
NH2 |
γ |
|
NH2 |
||
|
(?) |
|
|
|
|
(?) |
|
|
|
H |
|
|
|
|
|
|
|
H |
|
|
|
1, 2-H |
|
|
β |
|
1, 2-H |
||
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
H |
|
+ |
|
|
|
|
|
|
|
+ |
|
NH2 |
|
|
|
NH2 |
|
|
|
|
NH2 |
|
|
|
|
|
|
|
|||
|
|
|
|
+ |
|
|
|
|
|
|
|
|
|
SCHEME 17 |
|
|
|
|
|
However, this explanation contrasts with conclusions drawn from the general mechanism of alkyl radical loss from the analogous ionized alkenyl methyl ethers, in which ˇ-cleavage was found to involve an appreciable additional reverse critical energy138. Thus, CID studies show that CH3CHD(CH3)CCH2OCH3Cž eliminates the terminal methyl group via two consecutive 1,2-H shifts and -cleavage of the resultant ionized enol ether, CH3CH(CH3)CHDCHOCH3Cž , instead of undergoing ˇ-cleavage of the initial DI, CH3CHž (CH3)CH2CHC OCH3 ($ CH3CHž (CH3)CH2CHDOC CH3). Parallel effects were previously reported for alkyl radical losses from DIs isomeric with ionized carboxylic acids [e.g. CH3CHž (CH3)CH2CC (OH)2]97,98. The barrier towards the apparently favourable ˇ-cleavage is intelligible in terms of frontier orbital considerations and reflects the preference for radicals to add to the ˛- or -position of conjugated cationic systems100,101.
In |
the |
case |
of the C3H5NH2Cž species, ˇ-cleavage of žCH2CH2CHDNH2C |
gives |
the |
same |
product ion as ˛-fission of CH2DCHCH2NH2Cž or -cleavage of |
CH3CHDCHNH2Cž , so these possibilities cannot be distinguished by CID experiments. Nevertheless, depending on size of the barrier towards ˇ-fission, it may be more consistent

5. The chemistry of ionized, protonated and cationated amines in the gas phase 233
to regard hydrogen atom elimination from C3H5NH2Cž as an ˛- or -cleavage, even if the dissociating species have higher energies than žCH2CH2CHDNH2C .
Larger ionized cycloalkylamines show reactions involving DIs which represent extension of the chemistry of their acyclic counterparts. Thus, metastable ionized cyclohexylamine and ionized 2-methylcyclopentylamine undergo the same fragmentations, as is shown by the identical145 mass-analysed ion kinetic energy (MIKE146,147) spectra of these species. This common behaviour was interpreted in terms of reciprocal hydrogen transfers involving DIs (Scheme 18)145. Note how the loss of both C2H5ž and C3H7ž (in the ratio 100:5) is again attributed to -cleavage of species (ionized enamines, in this system) accessible by reciprocal hydrogen transfers. In contrast, the isomeric ionized cyclopentylmethylamine displays a distinct MIKE spectrum because ring opening to give similar stabilized DIs is no longer possible.
+ |
+ |
+ |
||||||
NH2 |
|
|
|
NH2 |
|
NH2 |
||
|
|
|
|
H |
1, 5-H |
|||
|
|
|
|
|
|
|
|
H |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
σ |
|
|
|
|
|
|
|
|
|
|
+ |
1, 4-H |
|
|
||||
+ |
+ |
|
|
|||||
|
|
|||||||
|
|
|
NH2 |
|
|
|||
NH2 |
|
|
|
|
|
NH2 |
||
|
|
|
|
|
+ |
|
|
|
|
|
|
|
|
|
|
|
+ C3 H7 |
|
|
|
|
|
||||
|
|
|
|
|
||||
+ |
|
+ |
|
|
|
NH2 |
|
NH2 |
+ |
||
|
|
|
|
|
NH2 |
H |
1, 4-H |
|
σ |
+ |
|
|
|
|
|
C2 H5 |
|
|
|
|
|
||
SCHEME 18
D. Reactions of Ionized Arylamines
It has long been known that the characteristic fragmentation associated with the ArNH2Cž entity is elimination of a neutral species having a mass of 27 a.m.u.2,3. Early results had also led mass spectroscopists to surmise that HNC, rather than the more stable tautomer, HCN, was eliminated. This conjecture was subsequently shown to be true by collision-induced dissociative ionization (CIDI) experiments, during which HNCCž is
distinguished from HCNCž148 . In this CIDI technique, the ionic fragment from dissociation of the parent ion in the field-free region of a mass spectrometer is removed by being retarded by an additional applied voltage148,149. The neutral fragment, which is not affected by this voltage, is then subjected to ionization and dissociation by collision with a target gas. The success of the CIDI experiments depends on the stability of HNC and

234 |
Richard D. Bowen |
HCN (and the corresponding ionized species) in the gas phase which arises because the facile intermolecular hydrogen transfers that would allow HNC to tautomerize to HCN in condensed phases are not possible. Thus, HNC is distinguishable from HCN because they may be specifically transformed to HNCCž and HCNCž , respectively, and these [H,C,N]Cž species dissociate in diagnostically different ways. Subsequent work, including high level MO calculations, indicates that both pairs of neutral and ionized isomers occupy energy wells; HCN is more stable than HNC (by ca 80 kJ mol 1), but the order of stability is reversed for the corresponding radical-cations (HNCCž is ca 75 kJ mol 1 more stable than
HCNCž ). Large barriers (125 275 kJ mol 1) exist towards interconversion of both neutral and ionized pairs by a 1,2-H shift150.
Similar experiments reveal that ionized N-heterocycles such as pyridines eliminate HCN, rather than HNC148. This distinction offers a potentially useful means of distinguishing between isomeric anilines and pyridines.
When ionized aminopyridines are considered, it is found that they resemble ionized anilines. Thus, all three isomers of C5NH4NH2Cž eliminate HNC; moreover, their distinct CID and CS spectra indicate that these three isomers of C5NH4NH2Cž do not interconvert before fragmenting151.
Some attention has been given to the structure of the C5H6Cž radical-cation formed by HNC loss from C6H5NH2Cž . It had long been tacitly assumed that this species was ionized cyclopentadiene. The energetics and kinetics of this reaction have been investigated by
photoionization, which indicates that the transition state lies ca 80 kJ mol 1 above the threshold for production of C5H6Cž and HCN152. Related studies on the pronounced dependence of the ionization energy for formation of C5H6Cž with the source residence times gave a similar transition state energy, which was interpreted to reflect formation of the less favourable combination of products, C5H6Cž and HNC, with little or no excess energy153. This view is consistent with the CIDI experiments148.
IV. PROTONATED AND ALKYL CATIONATED AMINES
A. Formation and Properties of Protonated and Alkyl Cationated Amines
Since most amines are basic, production of the conjugate acids by chemical ionization (CI)154,155 under proton transfer conditions is normally facile. Conversely, ammonia, methylamine and other small amines are often used as CI reagent gases (NH4C , CH3NH3C and related alkylammonium ions are formed as reagent ions). Variation of the reagent ion allows the exothermicity of protonation to be altered, thus permitting control and selectivity in the ionization process. However, many substrates react by capturing the ammonium ion, instead of abstracting a proton from it. The [MCNH4]C and related adducts which result from this reagent ion capture are useful in analysing numerous classes of compounds.
In contrast to protonated primary, secondary and tertiary amines, tetraalkyl ammonium ions, R4NC , obviously cannot be produced by protonation of the parent amine. In principle, however, these species may be made by alkyl cationation of the amine. In practice, other ionization methods are usually employed to form the gaseous ions from the appropriate salts. Thus, fast atom bombardment (FAB)156,157 of a solution of tetraalkylammonium bromides in a glycerol matrix is a convenient means of generating the free tetraalkylammonium ion in the gas phase. A modified procedure, in which an amine is dissolved in thioglycerol/2,20-dithioethanol matrix saturated with oxalic acid, allows RNH3C , R2NH2C and R3NHC ions to be produced under closely similar conditions158.
The advent of methods for determining proton affinities by studying bimolecular reactions in the gas phase has provided a wide range of interesting thermochemical data.