

12. The electrochemistry of the CDC, CDO and CDN groups |
621 |
Oxidation of the unsaturated tosylhydrazones 36 affords a mixture of |
ring-cleaved |
(38) and hydrolyzed (39) products explained in terms of a common
(Scheme 6)42. The formation of 38 and 39 can be intermediate (37). Oxidation of acylhydrazides (40)
follows an apparently mechanistically similar course to afford cyclized materials43 except in the presence of cyanide, in which case nitriles are formed44 (Scheme 7).
|
|
NHTs |
|
|
|
+ |
Ts |
|
|
N |
|
|
|
N |
|||
|
|
|
|
|
|
N |
|
|
|
|
R |
−2e− |
|
|
|
R |
|
|
|
|
−H+ |
|
|
|
|
|
(36) R = H or Me |
|
|
|
(37) |
H2 O |
|
||
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
H2 O |
+ |
|
O |
|
|
|
|
|
N |
N Ts |
|
|
|
|
|
|
|
||
|
|
R |
|
|
|
|
|
R |
|
(39) |
|
|
|
|
|
(37) |
|
|
|
|
|
|
SCHEME 6 |
|
||
R′ |
H |
|
|
|
|
R′ |
|
|
|
|
|
−2e− |
|
|
+ |
|
|
|
N |
|
R′′ |
|
|
R′′ |
||
R |
|
−H+ |
|
|
N |
|||
N |
|
|
R |
|
N |
|
||
|
(40) |
O |
|
|
|
CN− |
O |
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
||
|
R′ |
|
|
|
R′ |
|
|
|
R |
CN |
|
|
R |
N |
N |
R′′ |
|
|
H |
|
|
NC |
|
|
|
|
|
|
|
+ |
|
|
|
|
|
|
|
|
|
|
|
O |
|
|
|
|
|
|
H |
|
|
|
|
MeOH
N Ts
N
R
HO
R
O
(38)
R′ N
N
R
O +
R′′
−H+
(R′ = H)
N N
R |
R′′ |
O
SCHEME 7
V.CARBONYL (C=O) COMPOUNDS
A.Cathodic Reduction
1. Aldehydes and ketones
Earlier studies on the electrochemical reduction of aldehydes and ketones have been summarized in several reviews1,29,45; the reader should refer to those reviews for background material to the discussion which follows. Nevertheless, a short outline of the cathodic behavior of these substances will be helpful.
622 |
Albert J. Fry |
One of the primary issues in the electrochemical reduction of any carbonyl compound is the nature of the electroactive species under a particular set of experimental conditions, of which the proton donor ability of the solvent is paramount. The behavior of diaryl ketones is best understood in this respect. In acidic media (pH < 5), the species undergoing reduction is the protonated carbonyl compound (41) and the reduction affords a neutral ketyl radical (42). Dimerization of the latter affords benzpinacols, which generally rearrange to benzpinacolones under the acidic reaction conditions. In neutral and weakly basic media (pH from 5 to 11), the neutral carbonyl compound is reduced to a radical anion (43), which is protonated rapidly; the resulting ketyl radical usually immediately accepts a second electron and proton, and the product is the secondary alcohol. Reduction in strongly basic or aprotic media affords 43, which dimerizes to a pinacol. Aryl alkyl ketones and aldehydes have been generally believed to exhibit similar behavior (but see next paragraph). Dialkyl ketones and aliphatic aldehydes are not reducible at all under aprotic conditions; the LUMO of these substances is apparently too high to be accessible. These compounds are however reducible in protic media, e.g. alcohol solvents. Under these conditions the electroactive species is probably an alcohol carbonyl hydrogen-bonded complex. These types of compounds are generally reduced to alcohols; the intermediate ketyl radical is apparently reduced to an anion (44) at the very negative potentials at which reduction must be carried out, and protonation of 44 to afford the alcohol is undoubtedly fast.
|
H |
OH |
|
O− |
|
OH |
|
|
O |
|
|
|
|||
R |
R′ R |
R′ |
R |
R′ |
R |
− |
R′ |
|
|||||||
|
(41) |
(42) |
|
(43) |
|
(44) |
|
Attention has turned recently to the mechanistic details underlying these processes. Probably the most significant development in this area in recent years is the discovery that the ketyl radical produced by electrochemical reduction of benzaldehyde in buffered neutral ethanol is harder to reduce than benzaldehyde itself46. All previous discussions had assumed that the ketyl radical would be reduced as quickly as it is formed, but fast scan cyclic voltammetry demonstrated the existence of a short-lived intermediate, apparently the ketyl radical. Computer simulation of the voltammograms showed that the radical dimerizes at a rate ca 106 M 1 s 1.
Many aliphatic aldehydes exist primarily as hemiacetals in alcoholic solvents. It has been well understood for many years47 that the actual reducible species, or ‘electrophore’, in such media is not the hemiacetal but rather the small amount of the carbonyl compound itself (actually a hydrogen-bonded complex; see above) present at equilibrium. Thus reduction is kinetically controlled; that is, the overall rate of reduction is governed by the rate of conversion of the hemiacetal to the aldehyde. More recently, this has been confirmed and studied for formaldehyde and acetaldehyde in water at different pH levels48 and the kinetics of the reduction process have been studied for glucose, galactose and lactose49.
Synthetically, the question of which product is formed from a given carbonyl compound, the pinacol or the alcohol, is obviously of great interest. We have already noted the critical importance of the proton-donating ability of the medium in this respect. Attention has turned to the effect of other variables on the pinacol/alcohol ratio. Formaldehyde can be reduced cleanly to ethylene glycol at an electrode composed of a special type of carbon, in a process of potential commercial significance50. Nonaka and coworkers have found that reduction of aldehydes proceeds more selectively to aldehydes at nickel Teflon silica gel composite electrodes51. Nonaka and coworkers have also shown that for aromatic

12. The electrochemistry of the CDC, CDO and CDN groups |
623 |
aldehydes in aqueous methanol this ratio is sensitive to stirring conditions52: the ratio is in the range 2 3 when the electrolysis solution is stirred magnetically, but 24 34 under ultrasonic stirring. Presumably, highly efficient stirring (ultrasonic irradiation) removes the ketyl radical from the electrode surface before it can undergo further reduction. It would appear desirable to study the effect of ultrasonic stirring on a number of other electrode reactions.
Several studies have been made of the effect of added metal ions on the pinacol/alcohol ratio. Addition of antimony(III) chloride in catalytic amounts changes the product of the electrochemical reduction of acetophenone in acidic alcohol at a lead electrode from the pinacol in the absence of added metal salt to the secondary alcohol in its presence53. Antimony metal was suspected to be an intermediate in the reduction. Conversely, addition of Sm(III) chloride to DMF solutions of aromatic aldehydes and ketones54 and manganese(II) chloride to DMF solutions of hindered aromatic ketones55 results in selective formation of pinacols in excellent yields. When considering these results one should keep in mind the fact that aromatic ketones tend to form pinacols in DMF even in the absence of added metal ions1,29,45.
A number of investigators have studied the possibility of effecting electrochemical Grignard-type reactions by reducing a mixture of an alkyl halide and a carbonyl compound in the presence of a metal salt (or, equivalently, in a cell containing a readily oxidizable metal anode, thus generating the metal ion as current is passed) (equation 1, Section I). While these reactions have frequently been successful, it is not always clear whether the chemistry involves reduction of the carbonyl compound or the alkyl halide, or indeed perhaps of the metal ion. Examples include reduction of bromoesters in the presence of ketones in a cell containing an indium anode to afford Reformatsky-type products56, reduction of a mixture of an allylic halide and ketone in the presence of a catalytic amount of zinc bromide57, similar reductions involving a zinc anode and a catalytic amount of Ni(II)(bipyridine)58, the zinc anode alone59 and a magnesium anode and a small amount of samarium(III) chloride60. The latter reaction is thought to involve Sm(II) as the reactive species; the reactions involving zinc species are thought to involve a highly active form of zinc metal electrodeposited on the electrode surface, except for the Ni(II)(bipyridine) chemistry, which probably involves a low-valent nickel species. It has been established that a highly active form of zinc prepared by reduction of Zn(II) chloride by sodium in liquid ammonia readily effects the condensation of allyl halides with ketones61. The conversion of a mixture of an aldehyde or ketone and the tribromide 45 to an isoprenylated derivative by electrolysis in a cell with zinc anode is interesting
Zn+2 + 2e−  Zn0
Zn0
Br |
|
Br |
Br CH2 |
|
|
||
Br |
|
+ Zn0 |
|
|
+ |
ZnBr2 |
|
|
|
||||||
|
|
|
|
|
|
|
|
|
(45) |
|
|
|
|
|
|
|
|
|
|
|
O |
|
R |
|
|
|
|
|
|
|
|
Br CH2 |
|
|
BrZn CH2 |
|
HO |
CH2 |
|
|
|
Zn0 |
|
R |
R′ |
R′ |
|
SCHEME 8
624 |
Albert J. Fry |
because the electrogenerated zinc effects both dehalogenation of 45 and addition of the resulting monobromide to the carbonyl group (Scheme 8)59.
Tanko has shown that electrochemical reduction of ketone 46 affords a radical anion which undergoes conversion to an open chain species (Equation 21)62. Ring-opening was shown to be reversible; for this reason, ketones such as 46 are unreliable probes for electron transfer processes. Likewise, electrochemical reduction of aromatic aldehydes proceeds selectively to the corresponding benzyl alcohols in DMF containing a trialkoxysilane63. However, this process is thought to involve a catalytic cycle involving pentacoordinate hydrosilane intermediates; it is not a true electrochemical reduction and in fact consumes only a small amount of current.
|
O |
|
O− |
|
e− |
− |
|
|
(21) |
||
C6 H5 |
46 |
||
|
|
|
C6 H5 |
(46)
Much of the development of new reactions in modern synthetic chemistry rests on trapping intermediates in known chemical reactions in order to force the reaction into new pathways. This idea has been used to expand the range of usefulness of the electrochemical reduction of carbonyl compounds. We have seen that these reductions involve a variety of reactive intermediates (41 44). Ketyl radical 42 and radical anion 43 can be trapped by added good radical traps, though it is not always clear which of these species is involved in a particular process. As a rough rule of thumb we may expect 43 to be involved in aprotic or highly basic media, and 42 to be involved under neutral conditions. Alkenes serve particularly well as trapping agents. Reduction of ketones in DMF at a carbon electrode in the presence of alkenes affords alcohols in fair-to-good yields (equation 22)64.
Intramolecular versions of this reaction result in cyclization, as in equation 2365 and equation 2466.
R2
O
|
+ |
R3 |
e − |
HO |
|
|
|
(22) |
|
DMF |
|
|
R |
3 |
|
||
R1 |
R2 |
|
R1 |
|
|
|
||
|
|
|
|
|
|
|||
|
|
|
|
|
OH |
Me |
|
|
|
|
|
|
|
Me |
|
||
|
|
|
|
|
|
|
||
|
|
|
e |
− |
|
|
Me |
(23) |
|
|
|
|
|
|
|
||
|
|
|
DMF |
|
|
|
|
|
|
|
|
|
|
H |
|
|
|
|
O |
|
|
OH |
|
|
|
|
|
|
e − |
|
|
|
|
|
(24) |
|
|
DMF |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
( |
) |
|
H |
( )2 |
|
|
|
|
|
2 |
|
|
|
|
|
|
Addition to a vinylsilane takes place regioselectively (equation 25)67. A trialkylsilyl group can be used in this way to direct ring formation so as to form a six-membered ring (equation 26) in opposition to the usual preference for five-membered ring formation in
12. The electrochemistry of the CDC, CDO and CDN groups |
625 |
radical-type cyclizations67. These reactions undoubtedly involve attack of a radical anion on the CDC bond.
O |
R3 |
R2 |
R3 |
|
|
+ |
|
|
|
e − |
HO |
(25) |
|
|
|
|
|
DMF |
|
SiMe3 |
|
|
|
|
|
|
|
|
||
R1 |
R2 |
|
SiMe3 |
R1 |
||||
|
|
|||||||
|
|
|
|
|
|
Me |
OH |
|
|
O |
SiMe3 |
|
e − |
|
|
(26) |
|
|
|
|
|
|||||
DMF
 SiMe3
SiMe3
The radical anion is probably the reactive intermediate in the reductive carboxylation of aromatic ketones in DMF in a cell containing a sacrificial aluminum anode (equation 27)68; conceptually, this is better seen as a nucleophilic attack of radical anion 43 on the electrophile CO2. It is not clear whether the electrochemical cyclizations which have been observed onto a benzene nucleus69 and onto a cyano group70 (equations 28 and 29, respectively) involve 42 or 43, inasmuch as the reactions are carried out in the presence of a proton donor, albeit a rather poor one. Reduction of 47 is temperaturedependent: the hydroxy ketone (48) is the major product at room temperature, but 49 is the major product at 65 °C. The authors suggest that 49 arises by dehydration of 48 to an ˛,ˇ-unsaturated ketone (50) and subsequent reduction of the latter. This seems unlikely; reduction of 50 should afford a hydrodimer7,8, not 49. More likely, 48 is probably converted directly to 49 at the higher temperature by electrochemical cleavage of the activated carbon oxygen bond71.
|
|
|
|
CH3 |
|
|
e |
−, CO2 |
|
|
|
ArCOCH3 |
ArCCO2 H |
||||
90−99% |
|||||
|
|
|
|||
|
|
|
|
|
|
OH
e−
i-PrOH
O
(27)
OH
H  Me
Me
(28)
O |
CN |
|
X |
O |
|
|
|
|
|
||
|
e− |
|
|
|
|
( |
)n |
|
|
|
|
i-PrOH |
|
( )n |
|
||
|
|
|
|
||
|
|
|
|
|
|
( )m |
|
|
( )m |
|
(29) |
|
|
|
|
(47) |
(48) |
X = OH |
|
(49) |
X = H |

626 |
Albert J. Fry |
O
X
 ( )n ( )m
( )n ( )m
(50)
2. Carboxylic acids and acid derivatives
Carboxylic acids cannot normally be reduced directly electrochemically1; they can however be reduced to aldehydes by electrolysis in an undivided cell containing tributylphosphine and methanesulfonic acid72. The conversion involves an interesting combination of anodic and cathodic reactions (Scheme 9).
|
|
|
|
|
|
|
|
|
O |
|
|
|
|
|
||
Anode: |
|
[O] |
|
|
|
|
|
+ |
|
|
||||||
Bu3 P |
|
|
|
R |
|
|
|
C |
|
|
PBu3 |
|
|
|||
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
RCO2 H |
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
(51) |
|
|
|
|
|
||||
Cathode: |
|
|
|
|
|
|
OH |
|
|
|
|
|
||||
|
2 e−, 2H+ |
|
|
|
|
|
|
|
|
workup |
||||||
51 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
R |
|
|
CH |
|
|
PBu3 |
|
RCHO |
|||||
|
|
|
|
|
|
|
||||||||||
(stable)
SCHEME 9
Aroyl chlorides undergo an interesting electrochemical reduction to afford 1,2- dibenzoyloxystilbenes (54)73 75. The reaction appears to involve reductive dimerization to benzil (52) (Scheme 10). The latter is readily reduced to a stilbenediolate (53). Reaction of 53 with unreacted aroyl chloride affords 54 (trans:cis D 2 : 1). Alkene 54 can be further reduced to a diphenylacetylene by electrolysis at a more negative cathode potential74. Benzoyl fluoride undergoes the same conversion to 5474. Voltammetric investigations and careful product analysis have revealed the fact that these reactions are actually more complex than suggested by Scheme 10. Typically, at least two voltammetric waves are observed74,75. The most positive is that of the acid chloride itself. A second wave is usually seen for the corresponding benzaldehyde. Small amounts of the corresponding aldehyde also accompany 54 in the preparative electrolysis. Other electrolysis products include the corresponding carboxylic acid and anhydride; these presumably arise by reaction of the acid chloride with traces of moisture in the solvent74,75. Electrolysis of thiophene- 2-carbonyl chloride in the presence of D2O and CD3CN gave products suggesting the intermediate formation of the benzoyl anion75b. Reduction of phthaloyl chloride affords 55 in 65% yield76. In this system, the enediolate intermediate is trapped intramolecularly (Scheme 11). On the other hand, electrochemical reduction of terephthaloyl chloride (para-substituted) and its derivatives affords linear polyterephthaloylenes in good yield (95% for the parent compound)77.
Reduction of aliphatic acid chlorides follows a roughly similar course78. That is, tetrameric products similar to 54 are the major products, together with small amounts of the corresponding aldehyde. The latter has been suggested to be formed from both acyl radical
12. The electrochemistry of the CDC, CDO and CDN groups |
627 |
|
2e − |
|
Ar |
||
|
|
|
−O |
||
ArCOCl |
|
e |
− |
||
|
|
|
−Cl− |
||
52 |
|
2e − |
|
||
54 |
|
|
|||
− 2 A rCO2 |
− |
||||
|
|||||
O
Cl e−
Cl |
− Cl− |
|
O
O
Cl
O−
−O
Cl
O
• |
|
|
|
ArCOCOAr |
|
|||
ArC |
|
O |
|
|
|
|
||
|
|
|
|
|
||||
|
|
|
|
|
||||
|
|
|
|
|
||||
|
|
|
|
|
|
|
(52) |
|
|
O− |
2 A rCOCl |
Ar |
OCOAr |
||||
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
ArCO2 |
Ar |
Ar |
|
|
|
|||||
(53) |
|
|
|
|
|
|
|
(54) |
ArC CAr
CAr
SCHEME 10
|
O |
O |
Cl |
|
|
Cl |
O |
|
O |
O |
Cl |
|
O |
2e−
O
O
Cl
O
O
O
(55)
SCHEME 11
and acyl anion intermediates78a. On the other hand, reduction of 1-adamantanoyl chloride in CD3CN (a good hydrogen atom donor but a poorer proton donor) affords the deuterated aldehyde in high yield, suggesting that the critical intermediate is the 1-adamantanoyl radical78c. Presumably dimerization of the radical is sterically inhibited, permitting the hydrogen atom abstraction to dominate. Electrochemical reduction of glutaryl chloride (56) affords a -lactone (equation 30)78b. This appears to suggest that the first intermediate, a radical anion formed by addition of one electron to the carbonyl group of 56, cyclizes faster than it can eject chloride. It would be interesting to know whether glutaryl bromide
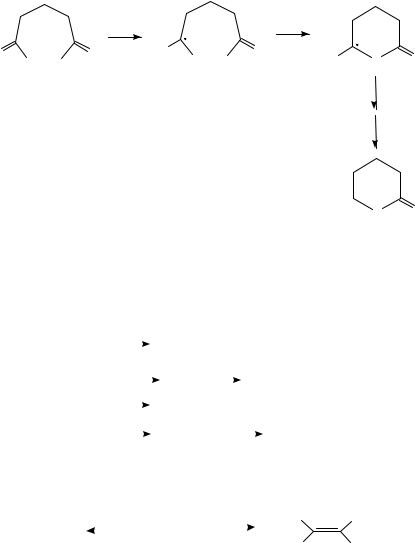
628 |
Albert J. Fry |
would take a different course because of the better leaving group ability of bromine.
|
|
e− |
|
|
|
|
O |
O |
−O |
O |
Cl |
O |
O |
Cl |
Cl |
Cl Cl |
|
|||
(56) |
|
|
|
|
|
|
(30)
O O
The electrochemical reduction of acid chlorides takes a very different course when carried out in an undivided cell equipped with nickel cathode and anode79. The product is a symmetrical ketone (57); 57 is formed by a complex sequence involving both electrodes (Scheme 12). This is really a chemical reaction induced by a highly reactive form of nickel produced by dissolution of the anode and plated onto the cathode. We have already encountered similar chemistry involving highly reactive zinc (Section V.A.1).
Anode: |
2e− |
|
|
|
|
|
|
|
|
|
|
|
|
Ni − |
|
|
|
|
Ni2 + |
|
|
|
|
|
|||
|
|
|
|
|
|
|
|||||||
Cathode: |
+ 2e− |
|
|
|
|
|
|
|
RCOCl |
|
|
||
Ni2 + |
|
|
|
|
Ni° |
RCONiCl |
|||||||
|
|
|
|
|
|
|
|||||||
|
|
|
|
+ 2 Cl− |
|||||||||
2 RCONiCl |
|
|
|
|
Ni2 + |
+ (RCO)2 Ni |
|||||||
|
|
|
|
||||||||||
(RCO)2 Ni |
|
|
|
|
RCONi |
|
|
|
R |
|
|
RCOR + Ni° + CO |
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
(57) |
|
|
|
|
|
|
|
CO |
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
SCHEME 12 |
|
|
|||||
ArCOCOAr |
e |
− |
|
e− |
Ar |
OSiMe3 |
||
|
|
|
ArCO2 R |
Mg anode |
|
|
||
Mg anode |
Me3 SiO |
Ar |
||||||
|
||||||||
|
|
|
|
|
Me3 SiCl |
|||
SCHEME 13
Esters undergo conversion to 1,2-diketones when reduced in a cell containing a magnesium anode80. Reduction in the presence of chlorotrimethylsilane affords an enediol bis-silyl ether (Scheme 13)80. Coordination of the chlorosilane probably activates the diketone for reduction80. The effect of the magnesium anode is less clear; coordination of magnesium ion to the diketone may stabilize it against further reduction; magnesium ions
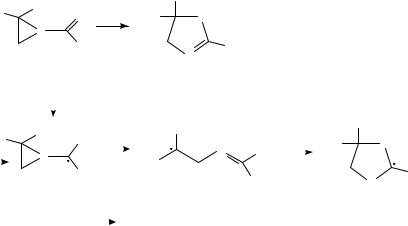
12. The electrochemistry of the CDC, CDO and CDN groups |
629 |
liberated at the sacrificial anode can presumably also facilitate reduction of the starting ester. This interpretation is bolstered by work of Pletcher and Slevin81, who showed that in DMF benzoate esters undergo alkyl-oxygen cleave to afford benzoate ion, whereas coupling is fast in the presence of added magnesium ion81. The role played by experimental conditions on these reactions still leaves much to be resolved. Thus, electrochemical reduction of esters and N, N-dialkyl amides takes a very different course when carried out in a t-BuOH/THF mixture in a cell containing magnesium anode and cathode; the ester is reduced to the primary alcohol stage82. Substitution of t-BuOD results in formation of RCD2OH from RCO2R0 . Ring-opening is observed with amide 5883, the isomerization of which to 61 is initiated electrochemically, but for which there is no net consumption of current (Scheme 14).
|
|
CH3 |
|
|
|
|
|
|
|
CH3 |
|
|
|
|
H3 C |
|
O |
|
H3 C |
O |
|
|
|
|
|||||
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
||||||
|
|
N |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
R |
|
|
|
N |
R |
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(58) |
|
|
|
|
|
(61) |
|
|
|
|
||
H3 C |
CH3 |
|
O− |
|
|
|
CH3 |
|
|
|
CH3 |
|||
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
N |
O− |
|
H3 C |
O |
||||||
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
N |
|
|
|
|
|
H3 C |
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
R |
|
|
R |
|
|
R |
||||
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
N − |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(59) |
|
|
|
|
|
|
|
|
|
(60) |
||
60 |
+ |
58 |
|
|
59 + |
61 |
|
|
|
|
||||
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
SCHEME 14
The electrochemical reduction of 2,6-pyridine diesters and S-thioesters shows interesting contrasts. The first step in each case is conversion to a short-lived radical anion, but the esters undergo alkyl-oxygen cleavage (cf Reference 81), whereas the S-thioesters undergo cleavage of the C(O) S bond84. The latter mode of cleavage is reasonable, inasmuch as S-thioesters resemble acid chlorides chemically more than they do esters.
3. 1,2-Dicarbonyl compounds
1,2-Diketones are easily reduced electrochemically because of the mutual electronwithdrawing effect of one carbonyl group on the other. Two-electron reduction usually takes place to afford an enediolate (62), which can then react with an electrophile E present in the medium (equation 31; see also Schemes 10 and 13); an alpha-hydroxy ketone is formed by subsequent tautomerization when E D H1,85,86. An unusual exception to the generalization that these substances undergo two-electron reduction is the electrochemical behavior of acenaphthenequinone (63), which is converted to the expected enediol dibenzoate (64) in the presence of aroyl chlorides but affords the dimeric substance 65 (shown to be the meso diastereomer by X-ray crystallography) in the presence of acetic
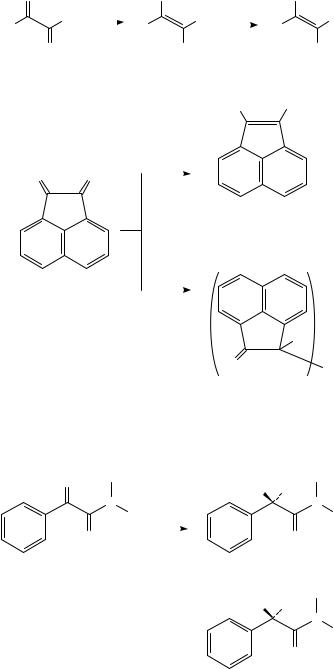
630 |
|
Albert J. Fry |
|
|
|
|
|
||
anhydride (Scheme 15)87. |
|
|
|
|
|
|
|
|
|
O |
|
|
O− |
|
|
|
|
OE |
|
R |
2 e− |
|
|
R |
|
2 E + |
|
|
R |
R |
|
R |
|
|
|
R |
(31) |
||
|
|
|
|
||||||
O |
|
|
O− |
|
|
|
|
OE |
|
|
|
(62) |
|
|
|
|
|
|
|
|
|
|
|
|
ArCO2 |
|
|
OCOAr |
|
O |
O |
|
2 e− |
|
|
|
|
|
|
|
A rCOCl |
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
(64) |
|
|
(63) |
2 e− |
|
A c2 O |
OAc
O
2
(65)
SCHEME 15
Schafer¨ examined the effect of chiral auxiliaries on the stereochemistry of reduction of a series of ˛-ketoamides (equation 32)88. Diastereomeric excesses ranged from 42 to 81%.
O |
R1 |
R1 |
||
|
|
HO |
H |
|
|
|
|
||
|
N |
N |
||
|
R2 |
R2 |
||
|
|
2 e− |
|
|
O |
|
2 H + |
|
O |
+ |
(32) |
|
R1 |
H |
OH |
|
N |
|
R2 |
|
O |