

Supplement A3: The Chemistry of Double-Bonded Functional Groups. Edited by Saul Patai Copyright 1997 John Wiley & Sons, Ltd.
ISBN: 0-471-95956-1
CHAPTER 12
The electrochemistry of the C=C, C=O and C=N groups
ALBERT J. FRY |
|
|
Wesleyan University, Middletown, Connecticut, USA |
|
|
Fax: (860) 685-2211; e-mail: AFRY@WESLEYAN.EDU |
|
|
I. INTRODUCTION . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
611 |
|
II. GENERAL CONSIDERATIONS . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
612 |
|
III. ALKENES (CDC COMPOUNDS) . . . . . . . . . . . . . . . . . . . . . . . . . . |
613 |
|
A. Cathodic Reduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
613 |
|
B. Anodic Oxidation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
614 |
|
C. Electroenzymatic Synthesis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
617 |
|
IV. CDN COMPOUNDS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
618 |
|
A. Cathodic Reduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
618 |
|
B. Anodic Oxidation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
620 |
|
V. CARBONYL (CDO) COMPOUNDS . . . . . . . . . . . . . . . . . . . . . . . . . |
621 |
|
A. Cathodic Reduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
621 |
|
1. |
Aldehydes and ketones . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
621 |
2. |
Carboxylic acids and acid derivatives . . . . . . . . . . . . . . . . . . . . |
626 |
3. |
1,2-Dicarbonyl compounds . . . . . . . . . . . . . . . . . . . . . . . . . . . |
629 |
B. Anodic Oxidation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
631 |
|
1. |
Aldehydes and ketones . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
631 |
2. |
Other carbonyl compounds . . . . . . . . . . . . . . . . . . . . . . . . . . . |
632 |
C. Electroenzymatic synthesis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
634 |
|
VI. ACKNOWLEDGMENTS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
637 |
|
VII. REFERENCES . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
637 |
|
I. INTRODUCTION
This review describes the electrochemical behavior of compounds containing the CDC, CDO and CDN functional group. The review covers both anodic oxidation and cathodic reduction of such compounds. The electrochemistry of these functionalities was reviewed
in an earlier volume of |
this series1; this article updates the previous one but does |
not include the material |
included there. The Kolbe oxidation of carboxylic acids has |
611

612 |
Albert J. Fry |
been adequately reviewed elsewhere2 and is not included here. This review also focuses primarily on reactions in which a compound containing a CDX (X D C, N or O) undergoes chemical transformation at an electrode, in distinction to processes wherein the CDX substance undergoes reaction with a reagent formed at an electrode. Thus we exclude from discussion here the cathodic hydrogenation of alkenes and alkynes at noble metal electrodes3 and aldehydes at carbon paste/palladium hybrid electrodes4; no matter how valuable it may be synthetically this reaction is basically a conventional surface catalytic process using electrochemically generated hydrogen. We also exclude a number of very useful and interesting processes5 in which the elements of X Y are added to a CDC bond through addition of an anodically generated electrophilic species ‘XC ’, followed by reaction with a nucleophile Y. Such processes do not involve the alkene itself at the electrode in any way. However, sometimes it is not easy to make this distinction; it may not be evident just what the actual electroactive species is in a particular transformation. This is particularly so in the case of electrochemically driven ‘Grignard-type’ processes, where cathodic reduction of a mixture of an alkyl halide, a carbonyl compound and an inorganic salt results in addition of the alkyl group to the carbonyl (equation 1). In principle, any one of the three components might be the electroactive species in such reactions. For this reason and also because this area has not been previously reviewed, a variety of such transformations are included herein even though some do not strictly speaking fit the criterion mentioned above.
|
O |
|
|
R′′ |
OM(n-1) |
|
|
|
|
||
|
+ |
R′′-Hal + M+n + 2e− |
|
|
(1) |
|
|
R′ |
|||
R |
R′ |
|
|
R |
II. GENERAL CONSIDERATIONS
The elementary electrochemical process exhibited by compounds containing the CDX functional group is electron exchange with the electrode: addition of one or more electrons to the LUMO at the cathode or removal of an electron from the HOMO at the anode, depending on whether one is carrying out a reduction or oxidation, respectively. Our knowledge from chemical experience of the dependence of LUMO and HOMO energies on structure will serve us well here. Thus we know that it is easier to add electrons to aldehydes and ketones than to alkenes because of the presence of the electronegative oxygen atom, and that alkenes bearing electron-withdrawing groups are more readily reduced and less readily oxidized than isolated alkenes. Likewise, conjugated species are more reactive toward both oxidation and reduction because the extended conjugation both lowers the LUMO energy and raises the HOMO energy relative to the corresponding unconjugated species. For these reasons, we may expect that for certain of the groups we discuss here cathodic reduction may be the predominant mode of electrochemical reaction; for others, the observed electrochemistry may be essentially completely anodic oxidation. In fact, with some exceptions, the chemistry discussed in this chapter involves the anodic oxidation of alkenes on one hand and cathodic reduction of carbonyl compounds on the other. Electrochemical reductions of alkenes and oxidations of carbonyl compounds are rare indeed.
The previous review in this series1 discussed the variations which can be expected in the structure and reactivity of electrochemically generated anion radicals and cation radicals as the group X in CDX is varied through C, N and O. The reader is referred to the previous review for that discussion, which will not be repeated here.
12. The electrochemistry of the CDC, CDO and CDN groups |
613 |
III.ALKENES (C=C COMPOUNDS)
A.Cathodic Reduction
Direct reduction of simple alkenes is difficult because their LUMO energy is very high. Normally, before a sufficiently negative electrode potential can be reached to effect electron transfer from the electrode to the alkene, some other constituent of the medium, such as the solvent or the supporting electrolyte, is found to be more easily reduced. Generally speaking, reduction of isolated double bonds is possible only under rather special conditions, for example, (a) electrolysis at a platinum or other noble metal electrode in a protic medium, in which case the reduction is effectively a catalytic hydrogenation4, or
(b) electrolytic reduction under conditions which favor ion-pairing, such as with a lithium salt as electrolyte and/or in a relatively nonpolar solvent6. In this case, electron transfer to the alkene to form the corresponding radical anion is made more thermodynamically favorable by ion-pairing of the cation of the supporting electrolyte to the incipient radical anion.
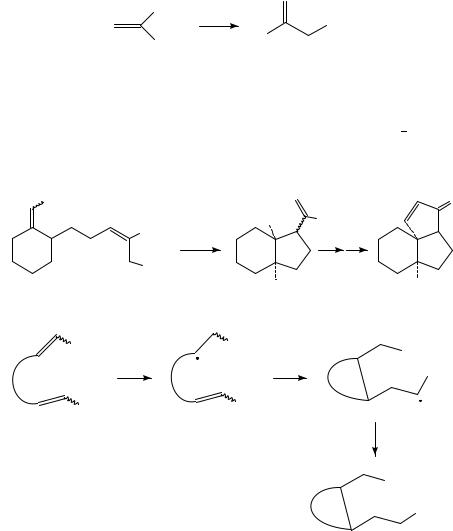
Reduction of conjugated CDC bonds is, however, much more readily accomplished because the LUMO energy of such substances is lowered considerably by conjugation. This is especially so in the case of alkenes bearing one or more strongly electronegative groups such as carbonyl, cyano, nitro etc., but the electrochemical behavior of such so-called ‘activated alkenes’ has already been adequately reviewed recently7,8. Such substituents are however not necessary: conjugated polyenes are also reducible at potentials which are readily accessible under electrochemical conditions. For example, reduction of 1,3-dienes in the presence of carboxylic esters results in cyclic alcohols (equation 2)9. To be sure, unusual experimental conditions are required: magnesium cathode and anode and a lithium perchlorate/THF solvent system. Almost certainly this reaction depends on strong ion-pairing of the diene anion radical to magnesium and/or lithium for its success.
R |
|
1. e− |
R |
|
|
+ |
R′′CO2 Me |
OH |
(2) |
||
2. H+ |
R′′ |
||||
|
|
R′
R′
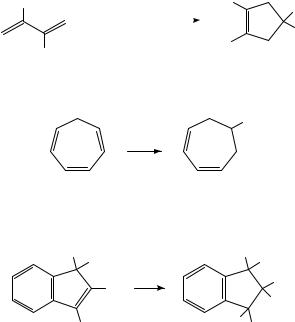
In the same vein, reduction of tropilidene (1) in the presence of butyl chloride results in reductive alkylation (equation 3). The reaction was employed in a synthesis of ˇ- thujaplicin10.
Bu
1. e |
− |
|
(3) |
2. H+
(1)
Farnia and coworkers have described a novel 1,2-phenyl shift which takes place during the electrochemical reduction of the indenes 2 (equation 4)11. Mechanistic experiments demonstrated that rearrangement takes place at the dianion stage.
C6 H5 |
C6 H5 |
|
C6 H5 |
H |
|
R |
C6 H5 |
|
|
(4) |
|
|
R |
|
|
|
|
C6 H5 |
H C6 H5 |
|
(2) R = H or Me
614 |
Albert J. Fry |
B. Anodic Oxidation
It is easier to oxidize an alkene electrochemically than to reduce it, because it is easier to reach powerfully oxidizing anode potentials12 (at which one can remove an electron from the HOMO of the alkene) without interference from the solvent or electrolyte than it is with reductions, where one can normally not achieve sufficiently negative cathode potentials to be able to add an electron to the LUMO. Even so, anodic oxidation of alkenes is relatively rarely observed; almost always the alkene is part of a conjugated system or it bears an electron-supplying substituent, which raises the HOMO energy.
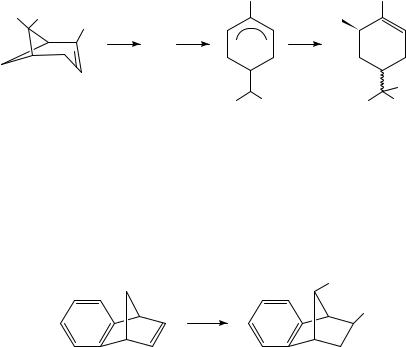
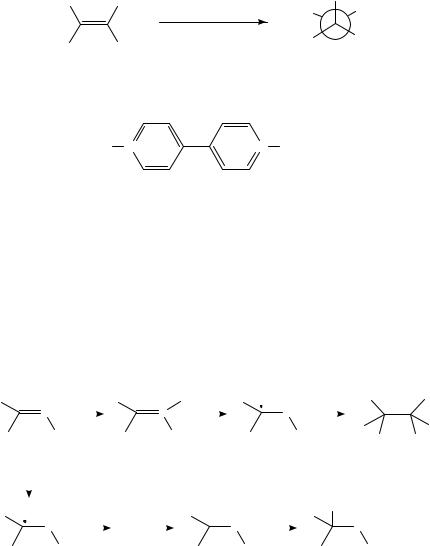
Oxidation of hydrocarbons, even alkanes and arenes, generally proceeds by removal of an electron from the hydrocarbon by the anode to afford a radical cation. Depending on the experimental conditions, the radical cation can react with a nucleophilic component of the medium or lose a proton or alkyl group. Either type of reaction affords a neutral radical. The radical is in general easier to reduce than the starting hydrocarbon because in the case of the radical the electron is being removed from a nonbonding molecular orbital, whereas with the starting hydrocarbon the electron being removed is in a bonding orbital. The product of this second electron transfer is a carbocation, which can undergo reaction(s) typical of such species. The oxidation of ˛-pinene (3) in methanol13 is typical of such oxidations (equation 5). The initially formed radical cation can relieve ring strain by bond cleavage to afford an allylic radical cation (4). Further oxidation of 4 and nucleophilic attack by the solvent lead to the observed products (5), which are stereoisomeric derivatives of the corresponding diol, sobrerol. Interestingly, a cathodic equivalent to the conversion of 3 to 5 has been observed previously14.
|
|
Me |
|
Me |
Me Me |
|
|
MeO |
|
Me |
|
|
|
|
|
• |
|
|
|
− e |
− |
|
|
|
|
3+ • |
|
|
(5) |
|
|
|
|
|
|
|
+ Me |
|
OMe |
|
Me |
Me |
Me |
|
(3) |
|
(4) |
|
(5) |
Anodic oxidation of 6 represents a variant on this scheme. Ring opening by cleavage of the bridge bond as observed for the radical cation of 3 does not take place. Instead, the 1,2- shift characteristic of these bicyclo[2.2.1] ring systems takes place to afford, ultimately, 7 stereospecifically15 (equation 6). Oxidation of tropilidene (1) affords the tropylium ion
(8)16; when the reaction is carried out in NaOMe/MeOH the product is tropyl methyl ether, which can be treated with acid to regenerate 8 or heated to induce rearrangement to 917. The latter can be oxidized further, followed by acid catalyzed hydrolysis, to afford tropone in good yield (Scheme 1). This work has been extended to a synthesis of 4-substituted tropones18.
OS
[O] |
OS |
|
(6) |
||
|
||
S−OH |
|
(6) |
(7) |
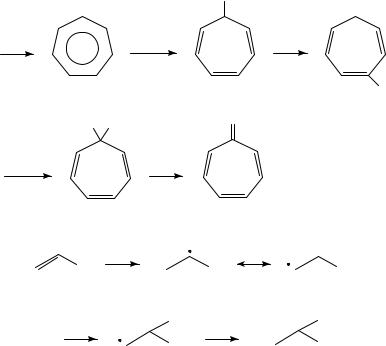
12. The electrochemistry of the CDC, CDO and CDN groups
|
|
|
|
|
OMe |
|
|
− 2e− |
+ |
|
|
NaOMe |
|
|
|
1 |
|
|
|
|
|
|
|
−H+ |
|
|
|
|
|
|
|
|
(8) |
|
|
|
|
|
(9) |
|
MeO |
OMe |
|
O |
|
|
|
|
|
|
|
|
|||
[O] |
|
|
|
H+ |
|
|
|
9 |
|
|
|
|
|
|
|
NaOMe |
|
|
|
|
|
|
|
MeOH |
|
|
|
|
|
|
|
|
|
|
|
|
SCHEME 1 |
|
|
|
|
−e |
− |
|
|
|
+ |
|
OR |
+ |
OR |
|
OR |
||
|
|
|
|
||||
|
|
|
|
|
|||
|
(10) |
|
|
|
(11a) |
|
(11b) |
|
|
|
|
|
Nu |
|
Nu |
11 |
Nu: |
|
|
|
|
+ |
|
|
|
|
|
OR |
OR |
||
|
|
|
|
|
|
||
|
|
|
|
|
|
|
(12) |
SCHEME 2
615
OMe
Anodic oxidation of alkenes is more readily accomplished when the double bond bears an electron-supplying group. Enol ethers, acetates and even a few enols have been studied. Removal of an electron from an enol derivative (10) affords a radical cation in which the major resonance contributor is presumably structure 11b, in which the positive charge is localized on the atom bearing the oxygen atom. Loss of a proton or alkyl group from 11 or reaction with a nucleophilic component of the medium leads to a neutral radical, which generally suffers further oxidation to a carbocation. Anodic oxidation thus converts the normally nucleophilic enol derivative 10 to at least two highly electrophilic species, 11 and 12. (Scheme 2). Synthetic applications can be designed to take advantage of this umpolung of reactivity of the enol derivative. Oxidation of enol acetates in the presence of a source of fluoride affords ˛-fluoroketones (equation 7)19. Two processes must take place: nucleophilic attack by fluoride and deacetylation, which probably also involves nucleophilic attack by fluoride on the carbonyl group. Plausible mechanisms may be written in which these two steps can take place in either order. Similarly, oxidation of cyclic enol derivatives in the presence of triphenylphosphine affords ˛-triphenylphosphonioketones (13), which can readily be converted into ˛-alkylidene ketones (14) (equation 8)20. On the other hand, oxidation of the dimesitylvinyl acetates 15 in the absence of an added nucleophile results in formation of benzofurans. It was suggested that the radical cation of 15 decomposes to an acetyl radical and cation 16. Intramolecular electrophilic attack on the adjacent arene ring, followed by methyl rearrangement, would lead to the observed products (equation 9)21. Evidence for this mechanism is the concomitant formation of biacetyl
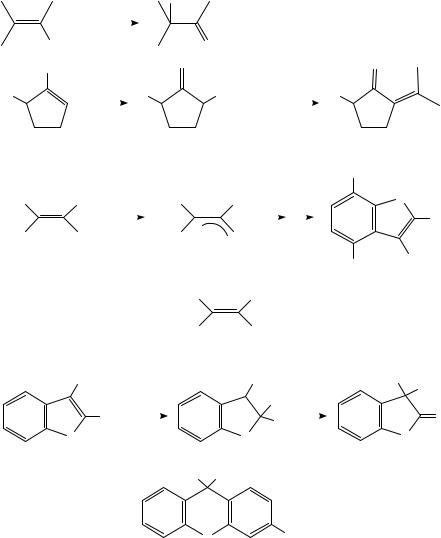
616 |
Albert J. Fry |
and the fact that the same benzofurans are also formed22 upon two-electron anodic oxidation of enolate 17; this should also afford 16. Benzofurans themselves are a type of enol derivative and Simonet has shown that they too undergo anodic oxidation. Two main types of products are formed: lactones (18) and xanthene derivatives (19) (Scheme 3)23. Attack of water on the oxygen-bearing carbon atom of the radical cation and a subsequent pinacol-type reaction involving migration of an aryl ring23 would afford 18. The origin of 19 is less clear; the authors suggested a phenoxonium ion as intermediate.
F
|
|
|
[O] |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(7) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
OAc |
|
|
Et3 N.3HF |
|
|
|
|
O |
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
OR′ |
|
|
|
|
|
|
|
|
|
|
O |
|
|
|
|
|
|
|
|
O |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
R |
|
|
[O] |
|
|
|
|
R |
P+(C6 H5)3 R′′CHO |
|
R |
|
|||||||||
( )n |
|
P(C6 H5)3 |
|
|
|
|
|
( )n |
|
|
base |
|
( )n |
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
(13) |
|
|
|
|
|
|
|
(14) |
(8) |
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Me |
|
Mes |
R |
|
|
|
|
|
Mes |
R |
|
O |
|||||||||||
|
|
|
|
|
−e |
− |
|
|
|
|
|
|
|
|
|
|
R |
||||
Mes |
OAc |
−A c. |
+ O |
|
|
||||||||||||||||
|
|
|
|||||||||||||||||||
|
|
|
|
|
Mes |
|
|
|
|||||||||||||
(15) |
|
|
|
|
|
|
|
|
|
|
|
(16) |
|
|
|
|
|
|
|
Me |
Mes |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(9) |
|
|
|
|
|
|
|
|
|
|
|
Mes |
R |
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
Mes |
O− |
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
(17) |
|
|
|
|
|
|
|
|
|
|
Ar |
|
|
|
|
|
|
|
+ Ar |
|
Ar |
Ar |
|||||||||
|
|
|
|
|
|
|
1. −e− |
|
|
OH |
|
|
|
||||||||
|
|
|
Ar |
|
|
|
|
|
|
|
|
|
|
|
O |
||||||
|
|
|
|
|
2. H2 O |
|
|
Ar |
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
O |
|
|
|
|
|
|
O |
|
|
O |
|||||||||||
|
|
|
|
|
|
3. −e− |
|
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(18) |
|
|
|
|
|
|
|
|
|
|
|
|
|
HO |
COAr |
|
|
|
||||||
O |
OMe |
|
|
(19) |
|
SCHEME 3 |
|
12. The electrochemistry of the CDC, CDO and CDN groups |
617 |
Barba and coworkers reported that the anodic oxidation of vinyl acetates in DMF affords alpha-formyloxy ketones (20) (equation 10)24. A novel feature of this reaction is the fact that the formyloxy carbonyl group of 20 is derived from the DMF solvent24.
O
R
[O] |
OCHO |
(10) |
|
DMF R
OAc
(20)
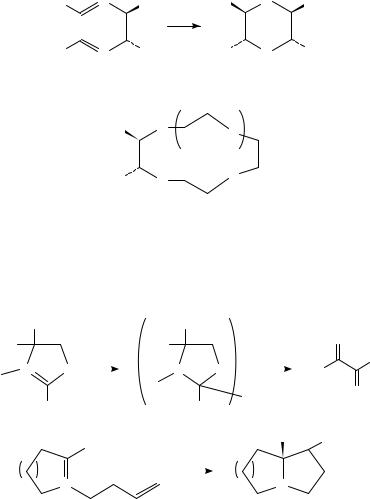
Moeller has carried out an extensive series of studies of the electrochemical oxidation of electron-rich bis-alkenes. One olefinic component is an enol ether, which is converted into an electrophilic center upon oxidation; this center then attacks the other site intramolecularly. The anodic oxidation of the bis-enol ethers 21 in methanol25 exemplifies the course of such reactions (Scheme 4). The products are bis-acetals (22), formed in 50 70% yield in many cases. The cyclization can be used to produce quaternary25 and angularly fused26 bicyclic and tricyclic structures (equation 11). In its original form, this work involved oxidation of a mono-enol ether bearing a nearby styrene-type double bond27.
OMe |
|
O |
|
|
(MeO)2 HC |
Me |
|
|
Me |
|
|
|
[O] |
|
|
|
MeOH |
|
|
|
SiMe3 |
|
|
|
H |
H |
|
|
|
(11) |
|
|
+ |
+ |
|
OMe |
OMe |
||
OMe |
|||
|
|||
|
|
||
|
−e− |
OMe |
|
OMe |
OMe |
|
|
|
|
||
(21) |
|
2MeOH −e− |
|
|
|
||
|
|
(OMe)2 |
|
|
|
(OMe)2 |
|
|
|
(22) |
|
|
SCHEME 4 |
|
C. Electroenzymatic Synthesis
Simon and collaborators have described a novel stereoselective electroenzymatic reduction of alkenes of type 23 (equation 12)28. The enzyme enoate reductase is the reductant
618 |
Albert J. Fry |
of the double bond in this process; the actual electroactive species is the methyl viologen dication (24) which is reduced at the cathode to the corresponding monocation, which then participates as an electrocatalyst in the enzymatic reduction process. This is representative of an increasing number of electroenzymatic processes which have been studied in recent years in an effort to combine the high degree of selectivity of enzymatic processes with the relatively low cost of electricity compared with that of redox coenzymes such as NADPH and NADH (see Section V.C).
R EWG
2e−, 2H+
MV+2 , enoate reductase
R′ |
R′′ |
(23)
|
H |
|
|
EWG |
|
R′′ |
|
R |
|
R′ |
(12) |
|
|||
|
H |
|
|
EWG = electron-withdrawing group
+ |
+ |
H3 C N |
N CH3 2 X− |
|
(24) |
|
IV. C=N COMPOUNDS |
A. Cathodic Reduction |
|
Electrochemical reduction of imines (25; Schiff bases) in acidic media proceeds via the iminium species, i.e. the protonated imine (26) (Scheme 5)29. Since 26 bears a positive charge, it is very easily reduced, so much so that the resulting neutral radical (27) is formed at a potential positive of its reduction potential. The products are therefore derived from 27 rather than the corresponding carbanion (28). This stands in contrast with the electrochemical behavior of imines in neutral media, where 27 is immediately reduced to 2830. Thus, cathodic reduction of bis-imines of 1,2-diamines (29) in DMF containing methanesulfonic acid affords tetrahydropyrazines (equation 13)31. A similar reaction can
R |
|
|
|
H+ |
|
R |
|
|
+ |
|
H |
e− |
R |
|
|
|
|
|
R |
R |
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
NH |
|
|
|
|
|||||
|
|
N |
|
|
|
|
|
|
|
N |
|
|
|
|
|
|
|
|
|
R′ |
R′ |
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
R′ |
|
R′′ |
|
|
|
|
R′ |
|
|
|
R′′ |
|
|
R′ |
|
R′′ |
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
RHN |
NHR |
||||||||||
|
(25) |
|
|
|
|
|
(26) |
|
|
|
|
|
|
|
(27) |
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
e− |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
R |
|
|
|
|
|
|
|
|
|
|
|
R |
|
|
|
|
|
R |
H |
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
N− |
|
H |
+ |
|
|
|
e |
− |
|
− |
|
|
|
|
|
|
||||||
|
|
|
|
27 |
|
|
|
|
NH |
|
|
|
|
|
NH |
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
R′ |
|
R′′ |
|
|
|
|
|
|
|
|
|
R′ |
|
|
R′′ |
|
|
R′ |
|
R′′ |
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(28) |
|
|
|
|
|
|
|
|
|
SCHEME 5
12. The electrochemistry of the CDC, CDO and CDN groups |
619 |
be used to prepare bis-azacrown ethers (30)32; the imine can be prepared in situ.
Ar N R′
2 H+
2 e−
|
H |
|
Ar |
N |
R′ |
(13)
Ar |
N |
R |
Ar |
N |
R |
|
(29) |
|
|
H |
|
|
|
|
|
|
|
|
|
H |
|
|
|
|
Ar |
N |
O |
|
|
|
|
|
|
n |
|
|
Ar |
N |
O |
|
|
|
|
H |
|
|
|
|
|
(30) |
n = 0− 4 |
|
|
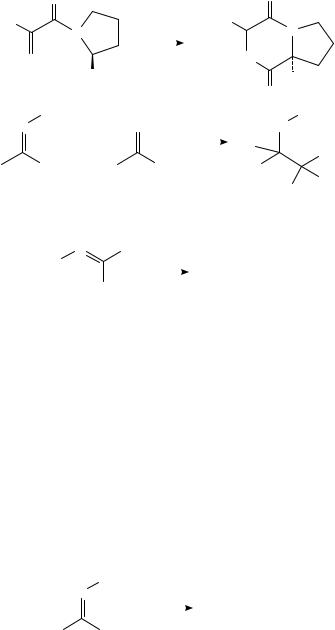
Reduction of N-alkyl iminium salts also ought to afford radical-derived products. Indeed, reduction of oxazolinium salts (31) results in isolation of the corresponding dimers, which can in turn be converted to the corresponding benzils by acid hydrolysis33 (equation 14). In a similar vein, electrochemical reduction of N-butenyl iminium salts (32) affords cyclopentane derivatives (equation 15)34.
R′ |
|
|
|
R′ |
|
|
|
|
|
R |
|
|
R |
|
|
|
|
O |
|
|
|
|
|
|
|
|
|||
+N |
|
e− |
|
|
|
H3 O+ |
|
Ar |
|
O |
|
N |
O |
|
|
Ar |
(14) |
||
|
|
||||||||
R′′ |
|
|
R′′ |
|
|
|
|
|
|
|
|
|
|
|
|
|
O |
||
|
|
|
|
|
|
|
|
|
|
|
Ar |
|
Ar |
2 |
|
|
|
|
|
(31) |
|
|
|
|
|
|
|
|
|
|
C6 H5 |
|
|
|
C6 H5 |
|
CH3 |
||
|
|
|
|
|
|
|
|
||
|
|
|
|
2e− |
|
|
|
|
|
n |
N |
|
H+ |
n |
|
(15) |
|||
ClO4 − |
|
|
|
N |
|
|
|||
+ |
|
|
|
|
|
|
|
|
|
(32)
It is known that electrochemical reduction of oximes in protic media occurs in two steps: the N O bond is first reduced to form an imine and the latter is then reduced to afford a primary amine1,29. Tallec has shown that the amine from oxime 33 can be trapped intramolecularly (equation 16)35. Interestingly, the SS diastereomer predominates; the chiral pyrrolidine ring derivative serves to control the stereochemistry of formation of the new benzylic chiral center. Electrochemical reductive cross-coupling of O-methyl oximes with carbonyl compounds in isopropanol at a tin cathode affords adducts (equation 17) which can be reduced further to 2-amino alcohols36. In this fashion, menthone could
620 |
Albert J. Fry |
be condensed with acetaldehyde O-methyl oxime to afford a chiral adduct which was effective as a promoter for the enantioselective addition of diethylzinc to aldehydes.
|
O |
|
|
|
|
|
|
|
|
O |
|
C6 H5 |
N |
|
|
|
− |
|
C6 H5 |
|
N |
|
|
|
|
|
4e |
|
|
|
|
(16) |
|||
|
|
|
|
|
|
|
|
|
|
||
|
NOH |
|
|
4H+ |
|
|
|
HN |
|
||
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
||
|
|
CO2 CH3 |
|
|
|
|
|
|
|
H |
|
|
(S)-(33) |
|
|
|
|
|
|
|
|
O |
|
|
OCH3 |
|
O |
|
|
|
|
|
OCH3 |
|
|
|
N |
|
|
|
2e− |
|
|
HN |
|
||
|
+ |
|
|
|
|
|
R′ |
|
|
||
R′ |
R′′ |
|
R′′′ |
|
i-PrOH |
R′′ |
(17) |
||||
R |
|
|
R |
|
|||||||
|
|
|
|
R′′′ |
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
HO |
|
Finally, reduction of 1,1-dichloroimines leads to formation of isocyanides in good yield (equation 18)37.
N Cl
R
Cl
B. Anodic Oxidation
2e |
− |
|
+ |
|
C− |
(18) |
|
|
|
R |
|
N |
|
||
−2 C l − |
|
|
|
||||
|
|
|
|
|
|
||
Compounds containing the CDN functional group derivatives undergo anodic oxidation when the nitrogen atom bears an electron-rich heteroatom. Perhaps the simplest such species are aldoximes, which are anodically oxidized to nitrile oxides (34)38. The reaction was carried out in an undivided cell39, hence the species 34 underwent immediate reduction to a nitrile (equation 19). However, since nitrile oxides are 1,3-dipolar species, one could in principle carry out the oxidation in a divided cell in the presence of a good 1,3-dipolarophile40 to effect the synthesis of substituted heterocycles.
RCH |
|
NOH |
[O] |
R |
C |
|
O |
[H] |
R |
C |
N |
19 |
D |
! |
C N |
|
! |
||||||||
|
|
|
|
|
|
|
|
|
||||
|
|
|
anode |
|
(34) |
|
cathode |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
Oxidation of silyl tosylhydrazones (35) in an aprotic medium (dichloromethane) also results in the formation of nitriles, but in this case the conversion is apparently a true anodic process41 in which the key difference from the anodic behavior of oximes is the presence of the p-toluensulfonyl group, which facilitates cleavage of the N N single bond (equation 20).
NHTs
N
[O]
|
|
|
R |
|
C |
|
N |
(20) |
|
|
|
|
|||||
|
|
|
||||||
|
|
CH2 Cl2 |
|
|||||
R |
SiMe3 |
|
||||||
|
(35) |
|
|
|
|
|
|
|