

26. N-Oxidative transformations of CDN groups |
1645 |
Nitrones, such as N-methylene-1-phenyl-2-propylamine N-oxide or N-benzylidene- 1-phenyl-2-propylamine N-oxide, have been found to produce so-called metabolicintermediate (MI) spectral complexes with phenobarbital-inducible P-450 characterized by Soret bands positioned around 455 nm54,136,137; the presence of NADPH/O2 was a prerequisite for adduct formation (Figure 2). Comparative studies on the correlation between MI complex formation and dealkylation of N-substituted phenylalkylamines suggested that the nitrones themselves were unlikely to represent the ultimate ligands, but rather served to release, upon their hydrolysis, primary hydroxylamines as precursors to the corresponding nitroso compounds presumed to bind to the heme iron of P-450136,137. A representative sequence of events for this process is given in equation 12. Iron chelation by the nitroso functionality affords a tight, quasi-irreversible complex associated with
severe inhibition of P-450 activity138.
|
|
|
|
|
O− |
||||||
|
PhCH2 CH |
|
|
|
|
|
|
|
|
||
|
|
N |
|
CHPh |
|
||||||
|
|
||||||||||
|
|
|
|
||||||||
|
|
|
|
||||||||
|
|
|
|
+ |
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Me |
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
|
N |
|
N |
||||||||
|
Fe |
|
|
|
|
|
|
|
|
|
|
N |
|
N |
|
|
|
|
|
|
P-450-Fe2+ |
||
|
|
|
|
|
|
||||||
|
N |
|
|
|
|
|
|
|
|
||
R |
|
|
O |
|
|
|
|
|
|
|
|
O
PhCH2 CHNHOH + Ph C
H
Me
(12)
+ PhCH2 CHN  O
O
Me
It seems noteworthy that, apart from the P-450 system, other hemoproteins also appear to be active in the production of nitrones from secondary hydroxylamines. Thus, in the presence of O2, hemoglobin brings about diarylnitrone (31) formation from N- benzylphenylhydroxylamine, presumably by a peroxidative mechanism139.
In addition to the pathways described above, nitrones can derive from condensation of a primary hydroxylamine with an aldehyde or ketone140. Under simulated biological conditions, Beckett and coworkers were able to demonstrate that primary hydroxylamines metabolically formed from amphetamine, mexiletine or norfenfluramine readily combined with ketones produced by metabolic deamination of the primary amines141. Reactions
can be formulated according to equation 13, |
where R1 |
D |
aryl, alkyl or aralkyl, and |
|||||||||||||
R2 D alkyl or H. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
O− |
|
|
|||
R1CH2 |
CHNHOH + |
R1CH2 C |
|
O |
|
|
R1CH2 CHN |
|
|
|
CCH2 R1 + H2 O |
|||||
|
|
|
|
|||||||||||||
|
|
|
|
|||||||||||||
|
|
|||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
+ |
|
|
(13) |
||
R2 |
R2 |
|
|
R2 |
|
|
|
|
R2 |
|||||||
Moreover, nitrones can be generated from N-hydroxy products during extraction into an organic solvent, such as diethyl ether. The latter contains acetaldehyde as an impurity even after careful distillation. Indeed, nitrones arising as artifacts from the reaction of primary hydroxylamines, metabolically derived from methamphetamine and chlorpromazine, with acetaldehyde upon sample treatment with ether have been identified23,39. Acetone is frequently used as one of the components of the solvent systems applied for TLC analysis.

1646 |
Peter Hlavica and Michael Lehnerer |
FIGURE 2. Difference spectrum produced during NADPH/O2-dependent rat liver microsomal metabolism of N-methylene-1-phenyl-2-propylamine N-oxide. The time interval between each scan is about 40 s. (Data taken from Ref. 136, with permission)
The presence of small amounts of this agent converts primary hydroxylamines completely to their nitrones141.
Further possibilities for nonenzymatic nitrone formation include the disproportionation of nitroxides in cases where the nitroxyl functionality is attached to an ˛-carbon bearing at least one hydrogen142 (equation 14), or the rearrangement of certain N-hydroxy compounds to the thermodynamically more stable tautomeric forms, as has been observed for the formation of ˛-aminonitrone from N-hydroxy-N-methylbenzamidine50 (equation 15).
H
|
N O. |
|
|
|
|
NOH + |
+ |
2 |
|
|
|
|
N O− |
||
|
|
||||||
|
|
|
|
|
|
(14) |
|
|
PhC |
|
NH |
|
|
PhC |
NH2 |
|
N |
|
|
|
|
N+ |
(15) |
|
|
|
|
|
|||
|
Me |
OH |
|
|
Me |
O− |
|
|
|
|
|
(39) |
|
||

26. N-Oxidative transformations of CDN groups |
1647 |
C. Enzymology of N-Oxide Formation
N-Oxygenation of heterocyclic aromatic amines to yield N-oxides appears to be a domain of the P-450 system, as the 4a-hydroperoxyflavin intermediate of the FMO has been recognized to be a not sufficiently strong oxidant to attack the nitrogen in this type of amines123. This view is consistent with the inability of highly purified FMO from rabbit liver to catalyze the formation of N-oxide 40 from pyridine29, while sensitivity of hepatic microsomal N-oxygenation of the base and some simple 3-substituted derivatives to the presence of CO, metyrapone, SKF 525A, n-octylamine and ethyl isocyanide suggests involvement in this process of P-45058,59,92,143. In accord with this, any treatment of microsomes causing destruction of P-450 also gives rise to a fall in pyridine N-oxygenase activity92. Moreover, immunochemical titration of microsomal fractions with antibody against NADPH-cytochrome P-450 reductase has been shown to inhibit pyridine N-oxide production, lending further support to a pivotal role of P-450 in catalysis59. The putative reaction sequence is presented in equation 16. A key step in heteroatom oxygenation is electron abstraction (a) to yield an aminium radical118. Electron transfer from this species (b) gives a cation/Fe3C DO pair that generates an Fe(III) N- oxide adduct (c). Indeed, N-oxidative turnover of pyridine by hepatic microsomes from phenobarbital-pretreated rabbits is associated with the gradual formation of a 442-nm- absorbing metabolic-intermediate spectral complex having the oxygen atom of the polar N O function coordinated to the heme iron29. This adduct decays to release stable N- oxide and ferric pigment (d).
[FeO]3 + + |
|
|
|
a |
[FeO]2 + + |
+ |
b |
Fe3 + |
|
O + |
+ |
: N |
|
|
|
. N |
|
|
N |
||||
|
|
|
|
|
|||||||
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
c |
|
(16) |
Fe3 + |
|
|
|
−O |
|||
|
|
|
+ |
||||
|
|
|
|
d |
Fe3 + |
+ |
|
|
|
|
|
N |
|||
O |
|
|
|
|
|||
|
|
|
|
||||
|
|
|
|
||||
|
|
|
|
|
|
|
|
N |
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|||
|
|
|
|
|
|||
Special attention has been drawn to the effects of selective P-450 |
inducers on |
||||||
the N-oxygenation of pyridines. Thus, pretreatment of animals with phenobarbital has been reported to result in a 3- to 13-fold increase in rates of N-oxide formation, while 3-methylcholanthrene left activities unaffected92. In rabbit hepatic microsomes, the barbiturate increased the level of immunoreactive P-450 4B, a low-affinity isoform (Km D 949 M) accounting for 80% of total pyridine N-oxide production at high amine concentration60. Pyridine administration to rats and rabbits substantially elevated the P-450 2E1 content of liver microsomes, whereas expression of P-450 4B was only marginally enhanced59,60. The former isozyme was recognized to represent a high-affinity pyridine N-oxygenase Km
Comparative studies60,93 with highly purified cytochromes P-450 1A2, 2B4 and 2E1 recombined with NADPH-cytochrome P-450 reductase and phospholipid revealed that the latter hemoprotein species catalyzed conversion of pyridine to its N-oxide with the largest turnover number (ca 5 min 1). It is interesting to note that P-450-enriched fractions,

1648 |
Peter Hlavica and Michael Lehnerer |
obtained from crude extracts of Streptomyces griseus, have been found to effect NADPHdriven N-oxygenation of pyridine when complemented with ferredoxin and NADPHferredoxin reductase from spinach144.
Participation of P-450 in the microsomal pyridyl N-oxygenation pathway has been further inferred for compounds such as cotinine145, metyrapone42 and 2-phenyl-1,3- di(4-pyridyl)-2-propanol30 from results obtained with diagnostic inhibitors or inducers. Cytochrome P-450 2B1, either heterologously expressed in human Ad293 kidney cells62 or highly purified from phenobarbital-induced rat liver63, preferentially attacks the pyridyl nitrogen in the pulmonary carcinogen NNK with Km D 131 M and a turnover number of about 0.3 min 1. Similarly, P-450 appears to be involved in the production of N-oxides from some benzopyridines, such as quinoline (41) or isoquinoline146.
Detailed work has been devoted to the elucidation of factors governing N-oxygenation of amino azaheterocycles. To this end, a series of substituted 2,4-diaminopyrimidines and 6-aminopurines was subjected to metabolic analysis8,16,64,65,147,148. Generally, P-450
 =
=
=
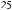
t 
t
τ
−
−
−
 −
−


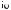



 τ
τ
FIGURE 3. Graphic displays of the cis and trans conformations of 9-benzyladenine (a) and variation of the potential energy of the azaheterocycle as a function of the torsion angles 1 and 2 (b). Contours are shown at intervals of 1 kcal mol 1 with the lowest energy level ( 17.6 kcal mol 1) indicated by the symbol (x). (Data taken from Ref. 148, with permission)

26. N-Oxidative transformations of CDN groups |
1649 |
was found to be responsible for oxidative attack at the vulnerable endo-nitrogens in these compounds to yield N-oxides (42, 43), as evidenced by blockage of this process by the presence of modifiers such as CO, SKF 525A, DPEA, metyrapone and n-octylamine8,16,148. The stimulatory action of pretreatment of animals with phenobarbital on N-oxide formation from the heterocycles was taken as an indication of the preponderant involvement of barbiturate-inducible P-450 isoform(s) in catalysis8,16,148. This type of isozyme is known to accommodate within its active site nonplanar nitrogenous compounds characterized by large depth, small area-to-depth ratio and flexibility in conformation149. Such a favorable geometry appears to be afforded by some 9-substituted adenines. Calculation of the potential energies as a function of bond rotations relating the 9-substituents with the ring system revealed 9-benzyl- and 9-benzhydryladenine to be low-energy conformers (Figure 3) most likely existing in the trans-form in solution65. Using 1H-NMR spectral techniques, it could be seen that the phenyl rings in those structures were close to the 8-H protons, exposing N 1 to permit 1-N-oxide (43) formation65. The N-oxy products, once formed, have been proposed to be stabilized by hydrogen bonding with the exo-amino group64. These findings were interpreted to provide evidence of the importance of stereochemistry in controlling N-oxygenation of these compounds.
Experiments with 5,6-substituted 2,4-diaminopyrimidines (pyrimethamine, metoprine) showed that N-oxygenation occurred at the ring nitrogens leading to the formation of both 1- and 3-N-oxides8. The failure to form any 1-N-oxide from 6-monosubstituted azaheterocycles obviously could not be due to steric hindrance by the substituent at the 6-position. In this case, amine imine tautomerism has been proposed to be a determinant of the site of biological N-oxygenation148,150, as illustrated by equation 17. Some correlation appears to exist between binding interaction of amino azaheterocycles with P-450 and susceptibility to N-oxygenation. Thus, the N-oxide forming 2,4-diamino-6-piperidinopyrimidine8 has been demonstrated to generate a marked low-spin adduct with ferric P-450148 characterized by a Soret band centered around 420 nm, while its 3-N-oxide derivative (19) elicited a 438-nm-absorbing spectral species151 with Kd D 2.4 M.
|
|
NH2 |
N |
NH2 |
|
|
|
|
N |
|
|
|
|
|
|
|
RH |
|
|
|
|
|
|
|
|
O |
(17) |
|
|
|
|
|
|
|
NH2 |
N |
NH2 |
|
NH2 |
N |
NH2 |
|
|
|||||
|
N |
|
|
|
N |
|
H |
|
|
|
|
|
|
|
R |
|
|
|
RH |
|
Electron density at a particular endo-nitrogen and lipophilicity are other factors which may have an influence on the N-oxygenation of pyrimidines and purines by the P-450
system64,65,147,148.
1650 |
Peter Hlavica and Michael Lehnerer |
VI. FURTHER TRANSFORMATIONS OF N-OXYGENATED
C=N FUNCTIONALITIES
A. Enzymatic Processes
1. Reductions
Among other options, N-oxygenated CDN functionalities can undergo enzymatic reduction. Thus, acetophenone oxime (23) has been found to be reduced under anaerobic conditions to the corresponding hydroxylamine by microsomes fortified with NADPH. The reductive process was sensitive to O2, but insensitive to CO and exhibited substantial species differences: rat liver homogenates reduced the oxime only to the level of the hydroxylamine, while liver preparations from guinea-pigs, mice and hamsters converted it to the amine152,153. Similarly, rabbit and human liver microsomes have been reported to reduce the amidoxime 25 derived from pentamidine to the parent compound with Km D 8.8 M; NADH was the preferred cofactor, and lowering the pH from 7.4 to 6.3 gave rise to a pronounced increase in the rate of reductive transformation, whereas the alternative substrate N-methylhydroxylamine was inhibitory51. The enzyme involved appeared to be identical with that responsible for the reduction of oximes 24, 26 and 27 originating from the N-oxygenation of benzamidines154, guanidines52 and aminoguanidines14, and seemed to correspond to the NADH-dependent reductase system previously purified from liver microsomes19,155.
Although heteroaromatic N-oxides are more stable metabolically than other types of N-oxides, there is ample evidence from experimental data that reduction does occur to a certain extent. When radiolabeled pyridine N-oxide (40) was administered intravenously to rats, almost 95% of the dose was recovered unchanged in the urine. However, after oral dosing, only 50% of the urinary radioactivity corresponded to the N-oxide, suggesting that the heteroaromatic N-oxide had been subject to reduction by the gut microflora156. This finding is in agreement with in vitro studies demonstrating the reduction of some pyridyl N-oxides by mammalian tissues. Thus, Chaykin and Bloch were the first to show nicotinamide N-oxide reduction by pig liver homogenates157. The same group succeeded to solubilize an enzyme from hog liver that reduced nicotinamide N-oxide to the parent amine. The reductase was a metalloflavoprotein and exhibited dependence on NADH158. This enzyme had properties similar to those of xanthine oxidase (XOD; EC 1.1.3.22), and XOD purified from milk was indeed able to catalyze the reduction of nicotinamide N-oxide; the reaction was inhibited by cyanide and oxygen159 and proceeded by direct transfer of the oxygen atom from nicotinamide N-oxide to xanthine160. In later experiments, evidence was provided that aldehyde oxidase (EC 1.2.3.1), supplemented with its electron donor, also functions as a major liver enzyme responsible for the reduction of nicotinamide N-oxide to nicotinamide161. It is interesting that the cytosolic aldehyde oxidase can mediate NAD(P)H-sustained nicotinamide N-oxide reduction when combined with the microsomal NADPH-cytochrome c(P-450) reductase (EC 1.6.2.4) redox protein162. Similarly, nicotinic acid N-oxide has been found to be reduced in vivo after an oral or intravenous dose to rats, and this activity was associated with the hepatic cytoplasm163.
Oral administration of 42 to rats resulted in extensive reduction to trimethoprim106. The 1,4-di-N-oxide olaquindox (49), a substance used as a growth promotor in cattle breeding, pig husbandry and poultry farming, has been shown to be converted to a limited extent to the 4-mono-N-oxide in rats164, and compound 46 was readily reduced to N,N- diallylmelamine both in vivo and in vitro109. The anerobic reduction, in the presence of xanthine oxidase, of a series of purine N-oxides, such as adenine 1-N-oxide or guanine

26. N-Oxidative transformations of CDN groups |
1651 |
3-N-oxide, has been pointed out by Stohrer¨ and Brown165. This metabolic step is considered a detoxification mechanism166 for this class of azaheterocycles, many of which exert oncogenic actions84.
O |
O |
|
CH2 OH |
|
||
|
|
|
|
|||
|
|
|
|
|
|
|
N |
C |
NHCH2 CH2 OH |
HOH2 C |
|
OH |
|
N |
Me |
|
N |
Me |
||
O |
|
|
|
O |
|
|
|
|
(49) |
|
|
(50) |
|
CH2 CHCOOH
N NH2
O
(51)
Microbial reduction of heteroaromatic N-oxides has also been reported. Thus, pyridine N-oxide (40) is reduced to pyridine in fermenting sucrose solutions containing baker’s yeast167. Similarly, the vitamin B6 derivative pyridoxine N-oxide (50) has been demonstrated to undergo reduction in yeast incubates168. Resting cells of Escherichia coli transform nicotinic acid N-oxide to nicotinic acid169; it has been concluded that certain microbes can adaptively produce enzymes which reduce the N-oxide, so that the nicotinic acid formed can be utilized. Pyridylalanine N-oxides (51) block the growth of Escherichia coli, the 4-isomer being the most potent inhibitor170. This isomer is the most rapidly reduced to 4-pyridylalanine, the putative toxicant. Since Lactobacillus arabinosus does not bring about reduction of the isomeric N-oxides, this organism remains unaffected by their presence170.
2. Oxidations
When 2,4,6-trimethylacetophenone oxime (23) was reacted with fortified rabbit liver supernatant, analysis of the extracts permitted the identification of the corresponding nitro derivative as a metabolite37. Similarly, incubation in the presence of NADPH/O2 of phenylacetone oxime (28) with hepatic preparations from rabbits, mice, hamsters, guinea-pigs and rats yielded variable amounts of 2-nitro-1-phenylpropane171. Formation of the nitro compound (52) was almost absent in assay media containing no cofactor or heat-denaturated supernatant, suggesting enzymatic turnover of the oxime172. The potential reaction sequence has been proposed to include N-oxygenation followed by isomerization172 (equation 18). The process is unlikely to be catalyzed by the FMO, since the 4a-hydroperoxyflavin does not attack oximes123. Matsumoto and Cho reasoned that oxidative activity might be associated with the cytosolic rather than with the microsomal liver fraction35.

1652 |
|
|
|
|
|
|
|
|
Peter Hlavica and Michael Lehnerer |
|
|
|||||||||||||
|
|
H |
|
|
O |
|
|
H |
|
|
O |
|
O |
|||||||||||
|
|
|
|
|
|
|
|
|||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O− |
|
|
|
+ |
PhCH2 |
|
C |
|
|
N |
|
PhCH2 |
|
C |
|
|
N |
|
|
|
PhCH2 CH |
N |
|||||||
|
|
|
|
|
||||||||||||||||||||
|
|
|
|
|
||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
+ |
|
|
|
|
|
O− |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Me |
|
|
|
|
|
Me |
|
|
|
|
|
|
Me |
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
(18) |
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(52) |
|
A novel pathway for oxidative transformation of oximes has been recently described. Thus, purified P-450 2C3 reconstituted with NADPH-cytochrome P-450 reductase and phospholipid has been found to convert the amidoxime derivative of pentamidine (25) to the corresponding amide and NO51. This isozyme also oxidized the guanidoxime (26) originating from N-oxygenation of debrisoquine to the urea derivative52. Reactions required the presence of NADPH/O2 and appeared to involve both the oxygenase and peroxidase activity of P-45051,52. In accord with these findings, a series of aldoximes, ketoximes, amidoximes and guanidoximes were oxidized by liver microsomes from dexamethasonetreated rats with the formation of nitrogen oxides, and it was concluded that oxidative cleavage of the CDNOH bond is a general P-450 3A-mediated reaction173 proceeding according to equation 19 (R1, R2 D H, alkyl, aryl, NH2). This process has been considered to be analogous to the biosynthesis of NO by two-step oxidation of L-arginine, as promoted by NO synthases (NOS, EC 1.14.13.39), which are hemoproteins related to cytochromes P-450174. The second step in the catalytic cycle, oxidative denitration of the guanidoxime intermediate (Nω -hydroxy-L-arginine) to release citrulline and nitric oxide, is likely to involve a Fe3C -OO species174 and is also brought about by the classical rat liver P-450175. Nitric oxide (endothelium-derived relaxing factor; EDRF) produced by the constitutive endothelial NOS inhibits adhesion of platelets and polymorphonuclear granulocytes to the endothelial surface and is an important factor in the maintenance and regulation of vascular tone176. It has to be mentioned that NO is capable of oxidizing guanidoximes to generate another potent, longer-lived, and as yet unidentified vasoactive agent, possibly HNO177.
R1R2C D NOH ! R1R2C D O C NO NO2 , NO3 |
19 |
3. Deaminations
Results on the role of oximes in deamination reactions are ambiguous. Phenylacetone oxime (28) was reported by Hucker and coworkers34 to be prone to enzymatic hydrolysis in rabbit liver supernatant to yield phenylacetone, the ketone being reduced to 1-phenylpropan-2-ol by the presence of microsomal oxidoreductases (equation 20). This mechanism requires incorporation of oxygen from water into the ketone. However, experiments with 18O in the atmosphere revealed that the carbonyl group of phenylacetone formed in rabbit liver microsomal incubates contained either 18O or 16O, the presence of 16O in the product obviously not arising from exchange of ketone 18O with solvent120. These findings were interpreted to mean that, apart from the hydrolytic route, a significant proportion of phenylacetone had been formed via an oxidative pathway, postulated as involving loss of ammonia from a carbinolamine intermediate120.
|
|
|
|
H2 O |
+ NH2 OH |
|
||||
PhCH2 C |
|
NOH |
|
PhCH2 C |
|
O |
(20) |
|||
|
|
|||||||||
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Me |
|
Me |
|
|
||||||

26. N-Oxidative transformations of CDN groups |
1653 |
Reinvestigation of the metabolic turnover of 28 in fortified hepatic microsomes from rabbits showed that the oxime did not readily undergo hydrolysis to ketone120. Similarly, acetophenone oxime has been demonstrated to be fairly resistant toward hydrolytic transformation in anaerobic rat liver homogenates152, while incubation of 2,4,6-trimethylacetophenone oxime (23) with rabbit liver preparations gave ketone and alcohol37. The stability toward hydrolysis of certain oximes on the one hand and the preponderant incorporation of 16O into the ketone products on the other led to the proposal that ketone formation might proceed via hydrolysis of the corresponding imine intermediates to eliminate ammonia87,120.
4. Isomerization and rearrangement reactions
As discussed previously, asymmetric oximes show geometric isomerism owing to restricted rotation about the CDN bond (equation 2). Investigations into the further metabolism of the Z-isomer of o-methylbenzophenone oxime, the predominant and more stable form of this N-oxy compound, in hamster hepatic microsomes disclosed conversion to the E-enantiomer, maximum transformation being dependent on the presence of NADPH, as was consistent with the participation in the isomerization process of microsomal enzyme(s)178. The extent of metabolic conversion of the Z- to the E-isomer was observed to vary with the species and sex of the animals used. The reaction was catalyzed by a number of organ homogenates, being maximal with lung and liver tissue. Microsomal isomerization was blocked by CO, SKF 525A and DPEA, indicating the involvement of P-450178. It was speculated that isomer interconversion took place via an ˛-hydroxy or a hydroxylamine intermediate.
Another interesting type of reaction is the oxime amide rearrangement, as has been reported for the enzymatic transformation of fluorenone oxime to phenanthridinone46 (equation 21). The process is catalyzed by NADPH-supported enzyme(s) located in the mitochondrial and microsomal fraction of rat liver, is insensitive to the presence of O2 and CO, but is stimulated by phenobarbital administration to the animals. The reaction mechanism has been postulated to be analogous to the Beckmann principle, the enzyme serving as an acid catalyst46. Similarly, n-butyraldoxime has been found to undergo a Beckmann-type dehydration catalyzed by P-450 to form butyronitrile179 (equation 22).
(21)
N O
N H
OH
PrCH |
|
NOH |
−H2 O |
PrC |
|
N |
(22) |
|
|
||||||
|
|
|
|
5. Conjugations
The OH-group in the E-configuration of acetophenone oximes has been proposed to be more accessible than that in the sterically hindered Z-conformers, and thus may be more

1654 |
Peter Hlavica and Michael Lehnerer |
susceptible to conjugation to yield water-soluble products85. Indeed, treatment with sulfatase of urine from rabbits dosed with 2,6-dimethylacetophenone imine caused a drastic increase in the amount of free acetophenone oxime (23) extractable, the E-form being the preponderant isomer85. These results indicated that rabbits excreted a considerable proportion of the isomeric ketoxime as a sulfate conjugate. Similarly, incubation with ˇ-glucuronidase or sulfatase of urine samples from rabbits administered amphetamine has been shown to be a prerequisite for detecting free phenylacetone oxime (28) as a metabolic derivative34. Moreover, evidence of the in vivo formation of conjugates from benzamidoximes (24) has been provided after hydrolytic cleavage180. Incubation of benzamidoxime with rabbit liver supernatant in the presence of uridine-50-diphosphoglucuronic acid (UDPGA) gave rise to a fall in the level of extractable free oxime86.
Until recently, N-oxides generated by oxygenation of aromatic heterocycles were regarded as metabolically stable, being excreted without further modification of the N O functionality. However, data have accumulated suggesting the N-oxide oxygen in such compounds to be a potential target for conjugation. Studies on the metabolic profiles of pyrimethamine and metoprine indeed showed that ˇ-glucuronidase treatment of urine from rats dosed with the diaminopyrimidines resulted in an increase in free 1- or 3-N- oxides, respectively, indicating that the N-oxides were at least partially transformed to glucuronic acid conjugates106. Similarly, the N-oxide drug minoxidil (19) has been found to be subject to glucuronidation, especially in monkeys and humans181,182.
The structures of the N-O-glucuronides produced from diaminopyrimidine N-oxides have not been fully established. There exist two possibilities, one of them implicating direct reaction of glucuronic acid with the N-oxide oxygen (equation 23a). Alternatively, amine imine tautomerism might afford an N-hydroxyimine configuration, followed by reaction of glucuronic acid with the N-hydroxy functionality (equation 23b). The latter mechanism has been favored by some investigators182. In this regard, it seems interesting that minoxidil can undergo sulfation directly at the N-oxide oxygen. The corresponding N-O-sulfate has been isolated from the bile of rats following intravenous administration of radiolabeled minoxidil183 and appears to be an active metabolite, since pretreatment of the animals with 4-acetamidophenol, a possible scavenger of sulfate groups, markedly decreased the hypotensive action of minoxidil183.
NH2 |
|
NH2 |
|
|
C |
O− |
C |
O glucuronide |
(a) |
|
N+ |
N |
+ |
|
(23)
NH |
|
NH |
|
|
C |
OH |
C |
O glucuronide |
(b) |
|
N |
|
N |
|
These findings were substantiated by in vitro experiments. Thus, rat liver cytosol has been detected to contain sulfotransferase (EC 2.8.2.1) activity which catalyzed