

Drug Targeting Organ-Specific Strategies
.pdf12.5 In Vitro Transport Studies |
319 |
The mechanisms of uptake and excretion of drugs by the liver has been widely studied using isolated hepatocytes and isolated perfused livers of rodents. The subject was extensively discussed by Oude Elferink et al. [82], summarized in a comprehensive special issue of the Journal of Hepatology in 1996 [83] and reviewed in several papers and chapters by our group [84,85]. In general, the rate and mechanism of drug uptake in isolated rat hepatocytes are very similar to those found in vivo. Only a few studies have investigated transport of drugs in human hepatocytes, and even fewer have used liver slices. However, studies in human and rat hepatocytes are hampered by the fact that the isolation procedures involve collagenase digestion for the disruption of cell-to-cell contacts. Clearly, this proteolytic enzyme may also damage plasma membranes and transport systems therein.
Jansen et al. [86] published an example of transport of a drug targeting preparation in human and rat hepatocytes. They found that the anti-viral drug ara-AMP coupled to lactosaminated human serum albumin, was taken up to the same extent by human and rat hepatocytes. This is one of the few examples where an equal rate of transport was found in both species. In general the uptake of drugs in human hepatocytes is slower than that in rat hepatocytes. In vit- ro–in vivo scaling calculations [87,88] showed that these differences in the uptake rate of drugs between rat and human cells, actually reflect inter-species differences rather than being due to differences in viability. Moreover, the differences described in rate and mechanism of drug transport in rat and man emphasize again that extrapolation to man of pharmacokinetic data obtained in rat, is hazardous. Most of the current knowledge on drug transport carriers is derived from experiments with rats. Therefore, more studies need to be carried out in human liver preparations to further elucidate the mechanisms of drug transport in the human liver and hence the relevance of animal data.
These results also indicate that human hepatocytes are an appropriate model in which to study inter-species differences and the mechanisms of hepatic transport in man.
In contrast to isolated hepatocytes, liver slices retain the cellular architecture of the liver without prior digestion with collagenase. This makes a systematic comparison of the data relating to transport of free drugs as well as drug targeting moieties from isolated hepatocytes and liver slices, an attractive model for studying the potential and limitations of the liver slice model in this area of research.
12.5.2 Transport in Liver Slices
Mechanisms of drug uptake in liver slices were studied in vitro as early as in 1963 by Schanker and Solomon [89]. The results obtained in these experiments are still valuable and show that the influence of temperature, anoxia, metabolic inhibitors and substrate inhibition can be successfully studied in this preparation. However, as mentioned before, at that time the preparation of reproducible precision-cut slices was not feasible. Therefore, the slice incubation technique was virtually abandoned in transport studies after the introduction of the successful isolation of rat hepatocytes.
In order to investigate the possibilities and limitations of the use of precision-cut liver slices prepared with a mechanical slicer in drug transport studies, different aspects of the mechanism of uptake of several classes of drugs in human and rat liver slices were investigated in our laboratory. Four model compounds which enter hepatocytes via entirely differ-

320 12 Use of Human Tissue Slices in Drug Targeting Research
Figure 12.4. Rhodamine B (25 M) distribution in the cross-section of a rat liver slice (± 250 M) after 5 min incubation. Fluorescence microscopy, bar = 100 M.
ent membrane transport mechanisms were investigated: the fluorescent dyes rhodamine B and lucigenin, the cardiac glycoside digoxin and the neo-glycoprotein lactosylated albumin. Receptor-mediated endocytosis into endothelial cells was studied with succinylated and aconylated albumin. The rate of penetration into the rat liver slice was studied with the lipophilic cationic compound rhodamine B (RB) and the hydrophilic organic cation lucigenin (LU). RB, which enters hepatocytes by passive diffusion was homogeneously distributed throughout the rat liver slice (250 m thickness) within 5 min [81] (Figure 12.4). These results indicate that for very lipophilic components both the penetration rate into the slice and the diffusion rate into all the cells are rapid processes. In contrast, after incubation with LU, which is taken up by hepatocytes through adsorptive endocytosis, fluorescence in the inner cell layers could only be detected after 15 min. If the rates of uptake of drugs in liver slices are to be compared with parameters obtained in vivo, some difficulties may arise. The uptake rate of compounds into the slice may not only reflect the uptake rate of the cells involved, but may also be influenced by the rate of penetration of the substrates (i.e. diffusion through sinusoidal spaces) into the slice. From our results with the lipophilic agent rhodamine B it is clear that the penetration process into the slice takes at least 5 min. Therefore, for substrates that are taken up into hepatocytes relatively fast, penetration into the slice may limit the uptake of the drug by the inner cell layers in the slice.
Digoxin uptake into rat liver slices showed a temperature-dependent component, compatible with the involvement of carrier-mediated uptake mechanisms. Quinine markedly inhibited the uptake of digoxin, in contrast to its diastereomer quinidine, which only slightly inhibited the digoxin uptake in rat liver slices. This stereoselective inhibition is in line with results obtained in isolated rat hepatocytes and isolated perfused rat livers [90,91]. These results were also found after cryopreservation of the slices, indicating that carrier-specific phenomena can be studied after cryopreservation [92].
From these results it can be concluded that liver slices are a powerful tool for studying the mechanisms and specificity of carrier-mediated uptake of drugs and drug interactions which occur at the transport level.
12.6 The Use of Liver Slices in Drug Targeting Research |
321 |
Many drugs that are taken up and metabolized by hepatocytes are excreted via the bile canaliculi into the bile. One of the remaining topics in liver slice research is the question of whether liver slices are capable of bile excretion via the bile canaliculus. Thompson et al.[93] showed that slices are capable of excreting bile acids, however, there is a need for more experiments to determine whether this excretion takes place across the bile canalicular membrane.
12.6 The Use of Liver Slices in Drug Targeting Research
12.6.1 Distribution and Transport of Drug Targeting Devices
In our Institute, drug targeting devices are predominantly developed for the treatment off inflammatory diseases of the liver, such as fibrosis and cirrhosis. In order to increase the therapeutic efficacy of drugs, human serum albumin is used as a protein backbone; modifying this protein by the attachment of different sugar groups or peptide molecules targets these modified proteins to specific cell types in the liver as described in more detail in Chapter 4. Antiinflammatory agents are subsequently coupled to the protein backbone to serve as effector moieties.
Further studies are needed to determine whether these liver-directed drug targeting preparations are actually delivered to the specific liver cells. In addition, experiments should be carried out to ascertain whether the drug coupled to a drug targeting device is released in the target cell in the liver, and to ensure that the drug is still active in these cells. Up until now the distribution of these modified proteins has been tested in vivo mainly in rats, and in vitro in the perfused rat liver or isolated liver cells (both parenchymal and non-parenchymal cells). Almost no studies have been performed in the target species, man. As the main aim of drug targeting research is the development of preparations for clinical use, we investigated whether precision-cut liver slices could be used to study the uptake of these modified proteins into target cells of the human liver.
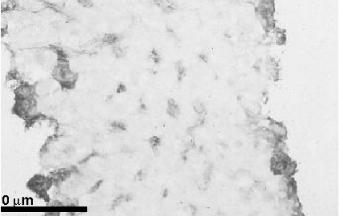
The temperature-dependent uptake and immunohistochemical localisation of modified proteins, Lactose27-Human Serum Albumin (Lact27-HSA), Succinylated-Human Serum Albumin (Suc-HSA) and Aconylated Human Serum Albumin (Aco-HSA), in rat and human liver slices showed that large molecules can enter the slice and are probably taken up by re- ceptor-mediated endocytosis (Figure 12.5). These large modified proteins were found distributed all over the liver slice, as was determined by immuno-histochemistry [81] (Figure 12.6). Recently, Beljaars et al. [94] showed that mannose-6-phosphate21-Bovine Serum Albumin (M6P21-BSA) is taken up by the non-parenchymal cells of human liver using slices from cirrhotic human livers. The non-parenchymal cells were identified as hepatic stellate cells (HSC) or endothelial cells. The involvement of hepatocytes, Kupffer cells, and bile duct epithelial cells was excluded. Until now, there has been little information concerning the presence of the mannose-6-phosphate receptor in human adult livers. The accumulation of M6P-modified albumin in human cirrhotic livers demonstrates, however, that M6P receptors are present in the human liver and that M6P-modified albumin may be useful as a drug carrier for the targeting of anti-fibrotic drugs in patients. In another study by Beljaars et al. [95],

322 12 Use of Human Tissue Slices in Drug Targeting Research
|
4 |
|
|
|
|
|
|
|
* |
factor |
3 |
|
|
|
|
|
|
|
|
Accumulation |
2 |
|
* |
|
|
|
|
||
|
* |
|
|
|
1 |
|
|
|
|
|
|
|
|
|
|
0 |
|
|
|
|
0 |
50 |
100 |
150 |
|
|
|
Time (min) |
|
|
4 |
|
|
* |
|
|
|
|
|
factor |
3 |
|
|
|
|
|
|
|
|
Accumulation |
2 |
|
* |
|
|
|
|
||
|
* |
|
|
|
1 |
|
|
|
|
|
|
|
|
|
|
0 |
|
|
|
|
0 |
50 |
100 |
150 |
|
|
|
Time (min) |
|
Figure 12.5. Uptake of 125I-Suc-HSA in liver slices from humans and rats at 37°( ) and 4°C
(•). The accumulation factor is defined as the concentration of the compound in the slices divided by the concentration in the medium. Each point is the mean of 5-6 separate experiments ± SEM. n = number of livers. *p < 0.05 versus 4°C. The dotted line represents the accumulation factor if 125I-Suc-HSA is exclusively distributed within the sinusoids.
the internalization of modified human serum albumin (HSA) with 10 cyclic peptide moieties recognizing the collagen type VI receptor (C*GRGDSPC*, in which C* denotes the cyclizing cysteine residues) yielding pCVI-HSA, was studied in normal and cirrhotic rat liver slices. 125I-pCVI-HSA was used to detect internalization in the liver slices. In contrast to 125I-HSA, an increase in the degradation products of 125I-pCVI-HSA was found over time, during the incubation of liver slices. These data show that pCVI-HSA is taken up and degraded in the cells of the liver slice. By immunohistochemistry it was shown that pCVI-HSA was specifically bound to rat HSC, in particular to the activated cells.
These distribution studies show that liver slices can be used to assess the level of uptake of a drug into the target cells in both healthy and diseased human liver. Isolation of non-
12.7 Efficacy Testing of the Drug Targeting Device in the Liver |
323 |
Figure 12.6. Fluoresceinlabelled aconylated human serum albumin distribution in the cross-section of a rat liver slice (± 250 M) after 120 min incubation. Bar = 100 M.
parenchymal cells from diseased livers is experimentally very difficult, therefore the liver slice technique offers a unique opportunity to study drug targeting preparations in diseased liver.
12.7 Efficacy Testing of the Drug Targeting Device in the Liver
It is of paramount importance to ensure that the drug targeting device with the drug attached or incorporated, is not only taken up by the target cells in the liver, but is also released from the device and remains active within the target cell. In the case of the development of drug targeting strategies for inflammatory liver diseases, an in vitro system was needed that could be used to test the anti-inflammatory effect of the drug-targeting preparations in the human liver.
To set up and validate the in vitro systems we initiated a study with rat liver slices. Stimulation by lipopolysaccharide (LPS) in liver slices was used to evoke a pro-inflammatory response in the liver. Lipopolysaccharide (LPS), a component of Gram-negative bacterial cell walls (also called endotoxin), has been associated with tissue injury and sepsis. In the liver LPS activates the resident macrophages, the Kupffer cells, which results in cytokine release [96]. Furthermore, LPS is cleared by the liver, mainly by Kupffer cells [97]. One of the major features of endotoxic shock is the induction of nitric oxide synthase in the liver [98]. Inducible nitric oxide synthase (iNOS), the expression of which is induced by LPS and cytokines, produces nitric oxide (NO) in large quantities [99].
The induction of iNOS by LPS, as observed in the hepatocytes in vivo [99], cannot be achieved in pure cultures of isolated hepatocytes. In fact, the induction of hepatocyte-associ- ated iNOS is found in pure cultures only after incubation with a mix of LPS and cytokines [100]. Induction of iNOS by LPS alone can be accomplished in co-cultures of hepatocytes and Kupffer cells with LPS [98]. These data indicate that induction of iNOS by LPS is mediated by cytokines released by the Kupffer cells in the liver and is thus a result of intercellular

324 12 Use of Human Tissue Slices in Drug Targeting Research
communications. However, the establishment of such co-cultures is technically complex while the cellular organization as present in the intact liver cannot as yet be achieved in culture. Therefore, to study the effects of LPS stimulation and the subsequent pharmacological intervention by targeted anti-inflammatory drugs in the whole liver in vitro, a system analogous to liver slices in which all cell types are present as in the original liver, would be the ideal.
Until recently only a few studies have focused on the activity or the viability of cell types other than hepatocytes within the liver slice [101,102]. Therefore, we attempted to establish whether non-parenchymal cells are still viable in the rat liver slice and whether they respond to LPS stimulation. Cytokine levels (tumour necrosis factor α (TNFα), interleukin 1β (IL-1β) and interleukin-10 (IL-10)) were measured in the incubation medium as a marker of non-parenchymal cell function. We found that in liver slices stimulated by LPS, IL-1β, TNFα and IL-10 are formed. In addition, the study was designed to elucidate the interaction between non-parenchymal and parenchymal cells in the liver after LPS induction. After LPS activation of rat liver slices iNOS was upregulated in the hepatocytes as determined by immunohistochemistry (Figure 12.7). This resulted in the production of NO, as measured by nitrate and nitrite (NOx) in the incubation medium (Figure 12.8). In addition to studying the pro-inflammatory response on a protein level, enzyme induction was studied at the transcription level. By RT-PCR, changes in specific mRNA were studied during the LPS-induced pro-inflammatory response. In rat liver slices the mRNA of iNOS was already upregulated only 2–3 h after induction with LPS, whereas the enzyme iNOS was found after 5 h.
Anti-inflammatory drugs such as pentoxyfilline and dexamethasone inhibited the release of cytokines and thereby the induction of iNOS and the release of NO in the LPS-stimulated liver slices.
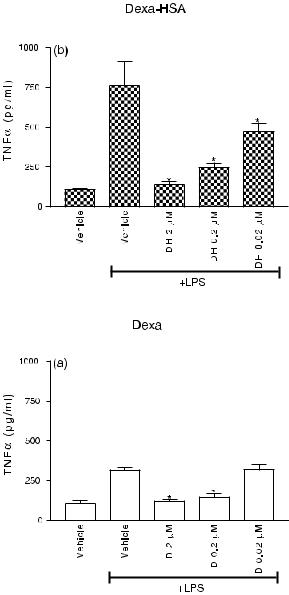
Melgert et al. [103] used this in vitro system to determine whether the drug targeting preparation containing the anti-inflammatory drug dexamethasone coupled to HSA, was still able to manifest its anti-inflammatory properties in liver slices. Dexamethasone10–HSA and uncoupled dexamethasone showed effective inhibition of LPS-induced NO and TNFα pro-
Figure 12.7. Cross-section of rat liver stained for iNOS. Left panel: control incubation after 24 h. Right panel: after 24 h incubation with 100 g ml–1 LPS.

12.7 Efficacy Testing of the Drug Targeting Device in the Liver |
325 |
|
80 |
|
|
|
|
|
|
|
|
|
|
|
* |
|
60 |
|
|
|
|
|
M |
|
|
|
|
|
|
x |
|
|
|
|
|
|
NO |
40 |
|
|
|
|
|
|
|
|
|
|
|
|
|
20 |
|
* |
|
|
|
|
|
* |
|
|
|
|
|
0 |
|
|
|
|
|
|
0 |
5 |
10 |
15 |
20 |
25 |
Time (hours)
Figure 12.8. NO production in rat liver slices after incubation in the absence or presence of 100 g ml–1 LPS for different time periods. The NO production is measured as nitrate/nitrite (NOx) concentrations in the medium ( M). Control (■) and + 100 g ml–1 LPS ( ). Data are expressed as mean ± SEM of four experiments. *p < 0.005 represents a significant increase in NO production by liver slices due to stimulation by LPS.
duction (Figure 12.9) in the liver slice model. These results show that the conjugate dexa- methasone10–HSA is taken up intracellularly and that active dexamethasone is released.
Studies similar to those described above are now being carried out in human liver slices. LPS induction in human liver slices also increased TNFα production to the same extent as was found in rat liver slices [104] (Figure 12.10). Human liver slices also produced IL-6, IL-8 and IL-1β, although the latter to a lesser extent than that observed in the liver slices of rat origin. However, human liver slices produced less NO after LPS stimulation than those of the rat. More experiments will be undertaken to elucidate this species difference.
Taken together these results indicate that non-parenchymal cells are still active in the slice preparations and that intercellular communication is still intact. Furthermore, pharmacological intervention by anti-inflammatory drugs can be successfully studied in liver slices. Together with the results obtained in regard to drug transport, liver slices seem to be a very promising in vitro system for studying intercellular distribution, cellular processing and effectiveness of anti-inflammatory drugs coupled to a targeting device.
326 12 Use of Human Tissue Slices in Drug Targeting Research
Figure 12.9. (a) TNFα production by rat liver slices (n = 9) after stimulation with 100 µg ml–1 LPS for 24 h in either the presence or absence of dexamethasone (D). Vehicle consists of PBS. *p < 0.05 versus vehicle + LPS. (b) TNFα production by rat liver slices (n = 9) after LPS stimulation for 24 h with or without Dexa10-HSA (DH). Vehicle contains PBS and an equimolar amount of HSA. *p < 0.05 versus vehicle + LPS.

12.9 Summary and Future Possibilities |
327 |
TNFα ng/ml
1.5
*
1
0.5
*
0
Human (n=4) |
Rat (n=4) |
Figure 12.10. Production of TNFα by human and rat liver slices in culture medium after 24 h stimulation with or without 100 µg ml–1 LPS. White bar: control; black bar: 100 µg ml–1 LPS present. Data are the mean of four experiments ± SEM. *p < 0.05 versus control.
12.8 Tissue Slices from Other Organs
Precision-cut tissue slices have also been prepared from other organs apart from the liver. Kidney slices are prepared by the same method as liver slices [73]. Kidney slices from different species, including man are used in the study of the toxicology and metabolism of drugs [65,66,73,105–108], organic anion and cation transport [109,110], release of prostaglandin and noradrenalin [36,111], and also in the study of organ preservation [70,112]. Since region-se- lective slices (cortex or medulla slices) can be prepared from the kidney, toxicity and metabolism in different regions of the kidney can therefore be studied [36,108,113]. Lungs cannot be sliced directly but need to be filled with 1.5% (w/v) low melting agarose solution containing 0.9% (w/v) NaCl at 37°C and allowed to gel on ice [73]. Lung slices have been used for drug transport and toxicity studies [114–118]. Up until now slices from other organs have not been used in the (transport) study of drug targeting devices, but like liver slices, these in vitro preparations have the potential and advantages to be useful in the study of transport, cellular processing and efficacy of drug targeting devices. In addition, slices of tumours could be used to study drug targeting in cancer research.
12.9 Summary and Future Possibilities
Drug targeting preparations are designed to be used in man, however most research with these preparations is carried out in animals. Due to known species differences, the study of these preparations in man in an early stage of development is therefore of paramount importance. In vitro studies exploiting human tissue can be used to ensure that these drug targeting devices reach the desired target cells and once there, are effective. When cells in the liver are the main target, in vitro research should be undertaken using preparations of both healthy and diseased human liver. As was discussed earlier in this chapter, liver slices seem like the ideal in vitro preparation for this purpose.The original architecture of the liver is still intact in the slice, which enables normal intercellular communication and cell-selective distribution of drugs. Slices can also be used to study drug interactions and the mechanisms and
328 12 Use of Human Tissue Slices in Drug Targeting Research
specificity of carrier-mediated uptake of drugs. In addition, the distribution of the drug into different cell types in the tissue can easily be studied in preparations of organ slices, as can the efficacy of the drug which is coupled to the targeting device. Furthermore, metabolism and toxicity of the drug targeting device or the released drug can be determined in the human liver. And finally, an important aspect of this type of in vitro research in man is, that it will ultimately lead to a reduction in the use of animal experiments.
In future, drug targeting devices aimed at other human organs may also be studied using precision-cut tissue slices.The latest data/literature on precision-cut tissue slices can be found at http://www.farm.rug.nl/slice/open.html.
References
[1]Trowell OA, Expert Cell Res. 1959, 16, 118–147.
[2]Warburg O, Biochem. Z, 1923, 142, 317–333.
[3]Krebs HA, Hoppe-Seyl. Z, 1933, 217, 190–227
[4]Stadie WC, Riggs BC, J. Biol. Chem. 1944, 154, 687.
[5]Krumdieck CL, Anal. Biochem. 1980, 104, 118–123.
[6]Seglen PO, Methods Cell Biol. 1976, 13, 29–83.
[7]Skett P, Toxicol. In Vitro 1994, 3, 491–504.
[8]Berry MN, Edwards AM, Barritt GJ, Isolated Hepatocytes Preparation, Properties and Applications, Vol. 21. Elsevier, Amsterdam, New York, Oxford, 1991.
[9]Guillouzo A, Oudea P, Le-Guilly Y, Oudea MC, Lenoir P, Bourel M, Exp. Mol. Pathol. 1972, 16, 1–15.
[10]Bojar H, Basler M, Fuchs F, Dreyfürst R, Staib W, Broelsch C, J. Clin. Chem. Clin. Biochem. 1976, 14, 527–532.
[11]Reese JA, Byard JL, In Vitro 1981, 17, 935–940.
[12]Groothuis GMM, Sandker GW, Pruim J, Weert B, Slooff MJH, Meijer DKF, Toxicol. In Vitro 1995, 9, 951–958.
[13]Dorko K, Freeswick PD, Bartoli F, Cicalese L, Bardsley BA, Tzakis A, Nussler AK, Cell Transplant. 1994, 3, 387–395.
[14]Moshage HJ, Rijntjes PJ, Hafkenscheid JC, Roelofs HM, Yap SH, J. Hepatol. 1988, 7, 34–44.
[15]Sandker GW, Isolated human hepatocytes: a research tool for investigations on drug transport, drug metabolism and liver transplantation. PhD thesis, University of Groningen, 1993.
[16]Ryan CM, Carter EA, Jenkins RL, Sterling LM, Yarmush ML, Malt RA, Tompkins RG, Surgery 1993, 113, 48–54.
[17]Schröder AJ, Blaheta RA, Scholz M, Encke A, Markus BH, Zentralbl. Chir. 1994, 119, 127–138.
[18]Takahashi M, Matsue H, Matsushita M, Nakajima Y, Uchino J, Artif. Organs 1993, 17, 653–659.
[19]Trevisan A, Gudat F, Guggenheim R, Krey G, Durmuller U, Luond G, Duggelin M, Landmann J, Tondelli P, Bianchi L, Hepatology 1982, 2, 832–835.
[20]Ballet F, Bouma ME, Wang SR, Amit N, Marais J, Infante R, Hepatology 1984, 4, 849–854.
[21]Houssin D, Capron M, Celier C, Cresteil T, Demaugre F, Beaune P, Life Sci. 1983, 33, 1805–1809.
[22]Hsu IC, Lipsky MM, Cole KE, Su CH, Trump BF, In Vitro Cell Dev. Biol. 1985, 21, 154–160.
[23]Miyazaki K, Takaki R, Nakayama F, Yamauchi S, Koga A, Todo S, Cell Tissue Res. 1981, 218, 13–21.
[24]Strom SC, Jirtle RL, Jones RS, Novicki DL, Rosenberg MR, Novotny A, Irons G, McLain JR, Michalopoulos G, J. Natl Cancer Inst. 1982, 68, 771–778.
[25]Tee LB, Seddon T, Boobis AR, Davies DS, Br. J. Clin. Pharmacol. 1985, 19, 279––294.
[26]Guillouzo A, Beaune P, Gascoin MN, Begue JM, Campion JP, Guengerich FP, Guguen-Guil- louzo C, Biochem. Pharmacol. 1985, 34, 2991–2995.
[27]Wiebkin P, Dees JH, Mathis JM, Prough RA, Drug Metab. Dispos. 1985, 13, 163–168.
[28]Blaauboer BJ, van-Holsteijn I, van-Graft M, Paine AJ, Biochem. Pharmacol. 1985, 34, 2405–2408.
[29]Friedman SL, Roll FJ, Anal. Biochem. 1987, 161, 207–218.
[30]Alpini G, Phillips JO, Vroman B, LaRusso NF, Hepatology 1994, 20, 494–514.
[31]Heuff G, Meyer S, Beelen RH, J. Immunol. Methods 1994, 174, 61–65.
[32]Zeitlin PL, Hubbard AL, J. Cell. Biol. 1982, 92, 634–647.