

Gallup G.A. - Valence Bond Methods, Theory and applications (CUP, 2002)
.pdf12.3 Dipole moments of CO, BF, and BeNe |
175 |
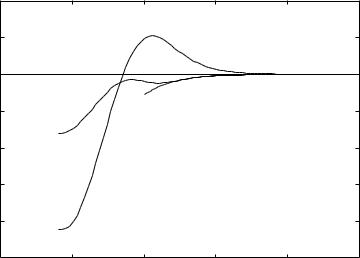
The curves for CO and BF show the form typical of these systems as is emphasized
in Ref. [53]. In our discussion of LiF in Section 8.3 we emphasized how the nature of a wave function could change from ionic to covalent with a change in internuclear
distance. Here again we appear to have the “signature” of this sort of phenomenon:
the change of sign of the moment at internuclear distances around 1.0–1.5 A |
|
|
|
|||
|
|
strongly |
||||
suggests the interplay of two effects where the winning one changes fairly rapidly |
|
|
|
|||
with distance. On the other hand the sign of BeNe does not change and this suggests |
|
|
|
|||
that one of these effects is absent in this molecule. |
|
|
|
|
||
From the signs on the moments and our work in Chapters 2 and 8 we interpret |
|
|
|
|||
these curves as follows (for the interpretation of the signs the reader is reminded |
|
|
||||
that all three of our systems are oriented with the |
less |
electronegative |
atom in |
the |
||
positive |
z-direction). |
|
|
|
|
|
1. At internuclear distances intermediate, but greater than equilibrium, the familiar ideas of |
|
|
||||
electronegativity win out, and the more electronegative atom has an excess of negative |
|
−0.70|e |. |
||||
charge. |
At the maxima the charge on O in CO is around |
−0.29|e | and o |
n F in BF |
|||
It is not surprising that in BF the effect is larger. No legitimate argument would suggest |
|
|
||||
that Ne has any sort of negative ion propensity, and we do not see a maximum in that |
|
|
||||
curve. |
|
|
|
|
|
|
2. When systems are pushed together, nonbonded electrons, on the other hand, tend to |
|
|
|
|||
retreat toward the system that has the more diffuse orbitals. In this case that is C, B, or |
|
|
||||
Be. Since the nonbonded electrons are generally in orbitals less far out, this effect occurs |
|
|
||||
at closer distances and, according to our calculations, wins out at equilibrium distances |
|
|
||||
for CO and BF. This is the only effect for BeNe, and the moment is in the same direction |
|
|
|
|||
at all of the distances we show. This retreat of electrons is definitely a result of the Pauli |
|
|
||||
exclusion principle. |
|
|
|
|
|
|
Both sorts of physical effects tend to fall off exponentially as the distance between |
|
|
||||
the atoms increases – the dipole moment must go to zero asymptotically. A close |
|
|
|
|||
examination of the CO results shows that the moment goes to very small negative |
|
|
|
|||
values again around 4.0 A |
|
|
|
|
|
|
. Whether this is real is difficult to decide without further |
|
|
||||
calculations. It might be that the Pauli exclusion effect wins again at these distances, |
|
|
||||
the result might be different for a still larger basis. Also, Gaussian basis functions |
|
|
||||
can cause troubles at larger distances because individually they really fall off much |
|
|
|
|||
too rapidly with distance. |
|
|
|
|
|
|
|
12.3.2 |
Difficulties with the STO3G basis |
|
|
|
|
We also calculated the dipole moment functions for CO, BF, and BeNe with an |
|
|
|
|||
STO3G basis, and it can be seen in |
Fig. 12.2 that there are real |
difficulties |
with |
|
|
|
the minimal basis. We have argued that the numerical value and sign of the electric |
|
|
|
|||




182 13 Methane, ethane and hybridization
constant. Using the methods of Chapter 5, we may write |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
T |
h R in terms of standard |
|||||||||||||||||||||
tableaux functions: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1 |
|
|
|
|
|
|
|
|
|
|
h |
|
|
|
|
|
|
|
|
|
|
h |
|
|
|
|
|
|
|
h |
|
|
|
|
||||||||||
T h R |
12 |
|
|
b |
|
|
1 s b |
|
6 |
|
b |
h |
z |
|
6 |
b |
2 pa x |
||||||||||||||||||||
|
|
|
|
|
|
|
|
h |
a |
|
|
1 s a |
|
|
|
|
|
|
|
|
h |
a |
1 s a |
|
|
|
|
|
|
|
h |
a |
1 s |
|
|
|
|
|
1 |
= |
|
|
|
h z |
|
|
|
− |
|
|
|
|
1 s b |
|
|
|
+ |
|
|
1 s b |
|
|
|
||||||||||||
|
5 |
|
|
|
|
|
5 |
|
|
|
5 |
|
|
||||||||||||||||||||||||
|
|
|
|
|
|
2 p x |
|
|
|
|
|
|
|
|
2 p x |
|
|
|
|
|
|
h |
z |
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
6 |
|
1 sa |
a |
|
1 sb |
b |
|
|
3 |
|
1 s a |
h |
z |
|
|
3 |
|
1 sa a |
2 p |
x |
|
||||||||||||
|
|
|
|
|
|
|
|
|
h |
|
|
h |
|
|
|
|
|
|
|
|
|
h a |
h |
b |
|
|
|
|
|
|
|
h |
|
h b |
|
|
|
|
|
− |
|
|
h z |
|
|
|
|
|
+ |
|
|
|
|
|
1 s b |
|
|
|
− |
|
|
1 s b |
|
|
|
||||||||||
|
|
5 |
x |
|
|
|
5 |
|
|
|
5 |
|
|
|
|||||||||||||||||||||||
|
|
|
|
|
|
|
|
2 p |
|
|
|
|
|
|
|
|
|
|
|
2 p x |
|
|
|
|
|
|
h |
z |
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
9 |
|
|
1 sa |
a |
|
h z b |
|
|
9 |
|
1 sa a |
2 pb x |
|
6 |
|
1 sa a |
2 |
zp |
x |
. (13.5) |
|||||||||||||
|
|
|
|
|
|
|
|
|
h |
|
|
1 s |
|
|
|
|
|
|
|
|
h |
1 s |
|
|
|
|
|
|
|
|
h |
h |
|
|
|
||
|
|
− |
|
|
h b |
|
|
|
|
|
+ |
|
h b |
|
|
|
− |
|
|
h b |
|
|
|
|
|||||||||||||
|
|
5 |
|
|
|
|
5 |
|
|
|
5 |
|
|
|
|||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
2 p x |
|
|
|
|
|
|
|
|
|
h z |
|
|
|
|
|
|
|
|
|
1 s b |
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
We see immediately that |
|
|
|
|
|
|
T |
1 |
AO , the standard tableaux function invariant to the hybrid |
|
|
|
|
||||||||||||||||||||||||
angles, is actually the largest term in |
|
|
|
|
|
|
T |
|
h R |
, but not overwhelmingly so. The others |
|
|
|||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||||
all depend on the hybrid angles and, therefore, so does |
|
|
|
|
|
|
1 |
|
|
|
|
|
T |
h R . We may also note that |
|||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
T 1h R |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1 |
|
|
|
|
|
|
|
|
|
|
using hybrid orbitals |
|
|
|
|
|
|
has a lower energy by |
|
|
|
≈3 eV, but, as seen from Table 13.1, |
≈3 eV. |
|||||||||||||||||||||||||
the full calculation, with either sort of basis, is still more stable by another |
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||
The calculated value of the dipole moment is 0.6575 D for this basis with the |
|
|
|
|
|
|
|||||||||||||||||||||||||||||||
charge positive at the H-atom end of the bonds. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CH |
3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Adding an H atom to CH |
|
|
|
|
|
|
2 |
|
might be expected to do little more than regularize the |
|
|
|
|
||||||||||||||||||||||||
hybrids we gave in Eq. (13.2), converting them to a canonical |
|
|
|
|
|
|
|
|
|
|
sp 2 |
set. With this we |
|||||||||||||||||||||||||
expect a planar doublet system. Whether the molecule is really planar is difficult |
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||
to judge from qualitative considerations. Calculations and experiment bear out the |
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||
planarity, however. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
A full valence orbital VB calculation in this basis involves 784 standard tableaux |
|
|
|
|
|
||||||||||||||||||||||||||||||||
functions, of which only 364 are involved in 68 |
|
2 |
|
|
|
|
2 |
A |
2 |
symmetry functions. For CH |
|
3 |
|||||||||||||||||||||||||
we present the results in terms |
of |
|
|
|
|
|
sp |
|
|
hybrids. This |
has |
no effect |
on |
the energy, |
|
|
|
||||||||||||||||||||
of course. We show the principal standard tableaux functions in Table 13.5. The |
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||
molecule is oriented with the |
|
|
|
|
|
|
|
|
C |
3 -axis along the |
|
z -axis and one of the H atoms on the |
|
|
|||||||||||||||||||||||
x -axis. The three trigonal hybrids are oriented towards the H atoms. The “ |
|
|
|
|
|
|
|
|
|
|
|
|
x ” sub- |
||||||||||||||||||||||||
script on the orbital symbols in Table 13.5 indicates the functions on the |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
x -axis, the |
||||||||||||||||||||||
“a ” subscript those 120 |
|
|
|
|
|
◦ from the first set, and the “ |
|
|
b ” subscript those 240 |
|
|
◦ from the |
|||||||||||||||||||||||||
first set. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
