

1. Кінетика хімічних реакцій у розчинах
Миттєва або істинна швидкість хімічної реакції за окремим компонентом за умови сталого об’єму системи визначається як зміна концентрації будь-якого учасника реакції i у певний нескінченно малий проміжок часу, тобто як похідна концентрації цього компоненту по часу:
![]() . (1.1)
. (1.1)
Швидкість
реакції завжди додатна. Якщо її визначають
за продуктом реакції
![]() ,
то в рівнянні (1.1) ставлять знак
,
то в рівнянні (1.1) ставлять знак![]() ,
якщо за вихідною речовиною
,
якщо за вихідною речовиною![]() – знак "–".
– знак "–".
Згідно
із законом діючих мас, швидкість
гомогенної реакції пропорційна добутку
концентрацій речовин, які вступають до
реакції, з певними показниками
степенів.
Вираз, що описує залежність швидкості
реакції від концентрацій реагентів,
називається кінетичним рівнянням
реакції. Якщо деяка реакція
![]() є простою (елементарною), тобто перебігає
в одну стадію, то кінетичне рівняння
має вигляд:
є простою (елементарною), тобто перебігає
в одну стадію, то кінетичне рівняння
має вигляд:
![]() , (1.2)
, (1.2)
де
![]() – константа швидкості реакції, яка
чисельно дорівнює швидкості процесу
за умови одиничних концентрацій
реагентів;a
і b
– частинні
порядки за компонентами А та В, відповідно.
Для простої реакції частинні порядки
мають такі самі значення, що й стехіометричні
коефіцієнти реагентів у рівнянні
реакції.
– константа швидкості реакції, яка
чисельно дорівнює швидкості процесу
за умови одиничних концентрацій
реагентів;a
і b
– частинні
порядки за компонентами А та В, відповідно.
Для простої реакції частинні порядки
мають такі самі значення, що й стехіометричні
коефіцієнти реагентів у рівнянні
реакції.
Якщо реакція, загальний вигляд якої наведено вище, є складною, тобто багатостадійною, то кінетичне рівняння набуває вигляду:
![]() . (1.3)
. (1.3)
У даному випадку показники степенів, до яких піднесені концентрації вихідних речовин, зазвичай не збігаються зі стехіометричними коефіцієнтами.
Загальний
порядок реакції
![]() –
це сума частинних порядків, а саме: для
рівняння (1.2)
–
це сума частинних порядків, а саме: для
рівняння (1.2)
![]() ,
а для виразу (1.3)
,
а для виразу (1.3)![]() .
Порядок реакції може бути цілим, дробовим
та нульовим. Частинні порядки іноді
набувають навіть від’ємних значень.
Порядок реакції не залежить від часу,
концентрацій реагентів (якщо немає
значного надлишку одного або декількох
з них) та температури. Він є формальною
ознакою, яка визначає ступінь залежності
швидкості реакції від концентрацій
учасників процесу.
.
Порядок реакції може бути цілим, дробовим
та нульовим. Частинні порядки іноді
набувають навіть від’ємних значень.
Порядок реакції не залежить від часу,
концентрацій реагентів (якщо немає
значного надлишку одного або декількох
з них) та температури. Він є формальною
ознакою, яка визначає ступінь залежності
швидкості реакції від концентрацій
учасників процесу.
Молекулярність є характеристикою простої реакції або окремої стадії складного хімічного процесу. Вона визначається числом частинок, що беруть участь у елементарному акті хімічної взаємодії. Існують моно-, бі- та (рідко) тримолекулярні реакції. Молекулярність не може бути більшою за три, оскільки одночасне зіткнення чотирьох та більше частинок є вкрай малоймовірним.
Для простої реакції порядок та молекулярність збігаються тоді, коли реагенти взято у стехіометричному співвідношенні. Якщо ж один з них міститься у надлишку, то молекулярність реакції не змінюється, а загальний порядок зменшується на величину частинного порядку за компонентом з надлишковою концентрацією.
Якщо складна реакція перебігає через ряд послідовних стадій, то її швидкість визначається швидкістю найповільнішої стадії, яку називають лімітуючою.
Константи швидкості реакцій різних порядків обчислюють за рівняннями:
![]()
![]() ; (1.4)
; (1.4)
![]()
![]() ; (1.5)
; (1.5)
![]()
![]() ; (1.6)
; (1.6)
![]()
![]() , (1.7)
, (1.7)
де
![]()
початкова концентрація вихідної
речовини; х
зменшення концентрації вихідної речовини
за час
початкова концентрація вихідної
речовини; х
зменшення концентрації вихідної речовини
за час
![]() ;
;![]()
поточна концентрація вихідної речовини,
яка відповідає часу
поточна концентрація вихідної речовини,
яка відповідає часу
![]() від початку реакції.
від початку реакції.
Порядок реакції за певним компонентом (частинний порядок) визначають на основі експериментальних даних про зміну концентрації цієї речовини з часом. Досліджуючи частинний порядок реакції за певним компонентом, всі інші реагенти беруть у надлишку. Внаслідок цього їх концентрації в ході досліду практично не змінюються і можуть бути включені до величини константи швидкості реакції. Таким чином, надлишок одного або декількох реагентів призводить до зменшення загального порядку реакції на суму частинних порядків за тими речовинами, концентрації яких є надлишковими.
Методи визначення частинних порядків поділяють на інтегральні та диференціальні. В інтегральних методах порядок визначають на основі експериментальних залежностей концентрацій речовин або інших, пропорційних концентраціям властивостей, від часу. В диференціальних методах застосовують залежності миттєвої швидкості за компонентом від його концентрації.
Для визначення частинного порядку реакції, наприклад, на основі методу підстановки, який належить до інтегральних, експериментальні дані послідовно підставляють у рівняння (1.4) – (1.7). Співвідношення, що дає приблизно однакові константи швидкості, які відрізняються у межах похибки досліду, вказує на порядок реакції за певним компонентом.
Часом
напівперетворення
![]() називається час, за який концентрація
вихідної речовини зменшується удвічі.
називається час, за який концентрація
вихідної речовини зменшується удвічі.
Внаслідок
підвищення температури швидкість
переважної більшості реакцій зростає.
Згідно з правилом Вант-Гоффа внаслідок
збільшення температури на 10 градусів
швидкість гомогенної реакції зростає
приблизно у 2 – 4 рази.
Температурний коефіцієнт швидкості
реакції
![]()
визначають як:
![]() , (1.8)
, (1.8)
де
![]() ,
,![]() ,
,![]() ,
,![]()
відповідно швидкості та константи
швидкості за температур, які відрізняються
на 10 К. У рівнянні (1.8) відношення швидкостей
реакції дорівнює відношенню її констант
швидкостей, оскільки концентрації
реагентів в результаті збільшення
температури на 10 градусів майже не
змінюються. Зазвичай у температурні
залежності швидкості реакції замість
неї підставляють саме константу
швидкості, оскільки вона, на відміну
від швидкості, не залежить від часу та
концентрацій.
відповідно швидкості та константи
швидкості за температур, які відрізняються
на 10 К. У рівнянні (1.8) відношення швидкостей
реакції дорівнює відношенню її констант
швидкостей, оскільки концентрації
реагентів в результаті збільшення
температури на 10 градусів майже не
змінюються. Зазвичай у температурні
залежності швидкості реакції замість
неї підставляють саме константу
швидкості, оскільки вона, на відміну
від швидкості, не залежить від часу та
концентрацій.
Якщо відомо константи швидкості для будь-яких двох температур, то
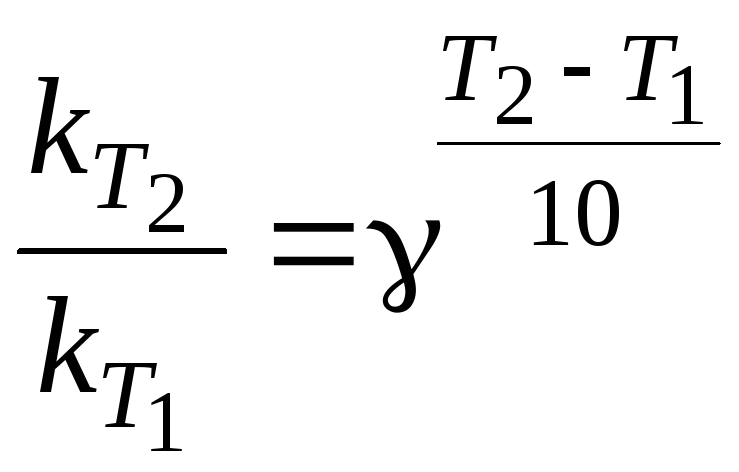 . (1.9)
. (1.9)
Температурний
коефіцієнт
![]() внаслідок зростання температури
зменшується, тому розрахунки за рівняннями
(1.8) та (1.9) мають наближений характер і
можливі тільки у неширокому температурному
інтервалі.
внаслідок зростання температури
зменшується, тому розрахунки за рівняннями
(1.8) та (1.9) мають наближений характер і
можливі тільки у неширокому температурному
інтервалі.
Значно краще залежності швидкості та константи швидкості реакції від температури описує рівняння Арреніуса, диференціальна форма якого має вигляд:
![]() , (1.10)
, (1.10)
а
інтегральна за умови
![]() :
:
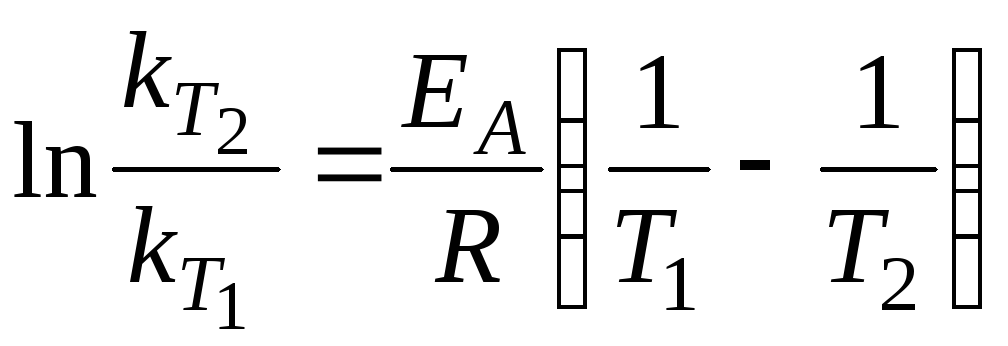 . (1.11)
. (1.11)
В
рівняннях (1.10) та (1.11)
![]() – енергія активації реакції, тобто та
мінімальна енергія, яку потрібно надати
1 молю вихідних речовин, щоб вони мали
змогу перетворитись у продукти.
– енергія активації реакції, тобто та
мінімальна енергія, яку потрібно надати
1 молю вихідних речовин, щоб вони мали
змогу перетворитись у продукти.![]() для певного хімічного процесу є сталою
величиною. ЇЇ можна змінити, тільки
ввівши в систему каталізатор. Енергію
активації обчислюють, визначивши
константи швидкості принаймні за двох
температур:
для певного хімічного процесу є сталою
величиною. ЇЇ можна змінити, тільки
ввівши в систему каталізатор. Енергію
активації обчислюють, визначивши
константи швидкості принаймні за двох
температур:
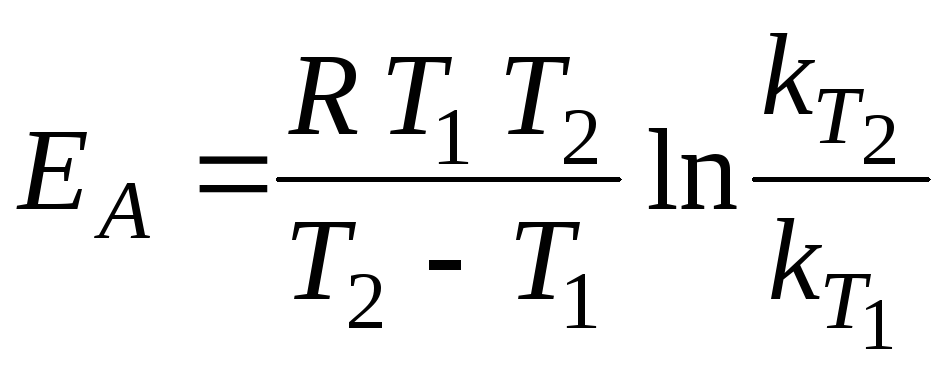 .
.
Інтегральну
форму рівняння Арреніуса (1.10) за умови
![]() можна записати також у вигляді:
можна записати також у вигляді:
![]() , (1.12)
, (1.12)
де
![]() – стала інтегрування. З виразу (1.12)
випливає, що
– стала інтегрування. З виразу (1.12)
випливає, що
![]() є лінійною функцією від
є лінійною функцією від
![]() .
Тангенс кута нахилу цієї прямої та
енергія активації пов’язані залежністю
.
Тангенс кута нахилу цієї прямої та
енергія активації пов’язані залежністю
![]() . (1.13)
. (1.13)
Для більшості гомогенних реакцій енергія активації лежить у межах 50 – 200 кДж/моль.
Робота 1.1. Дослідження кінетики розкладу пероксиду водню у водному розчині за сталої температури.
Мета роботи: визначити газометричним методом порядок реакції розкладу пероксиду водню у водному розчині та дослідити вплив концентрації каталізатора КІ на швидкість цього процесу.
Пероксид водню у водному розчині самочинно повільно розкладається за рівнянням:
2Н2O2 = 2Н2O + О2 . (1.14)
Наявність у розчині деяких сполук, які є каталізаторами, значно прискорює цей процес.
За перебігом реакції спостерігають, вимірюючи через певні проміжки часу від початку реакції об’єм кисню, що виділяється (газометричний метод). Схему приладу для дослідження кінетики реакції (1.14) зображено на рис. 1. Порівняльну лійку 4 спочатку закріплюють у штативі так, щоб рівень води у газовій бюретці 3 був на 2 – 2,5 см вище за позначку "0".
В окремі коліна реакційної пробірки 1 наливають піпетками зазначені викладачем об’єми розчинів пероксиду водню та каталізатора – водного розчину KI. Щільно закривають пробірку пробкою, яка сполучена гумовою трубкою з газовою бюреткою. Рівень води у газовій бюретці незначно знизиться.
Пробірку занурюють у кристалізатор з водою 2, який виконує роль термостата. Нахиляючи пробірку, змішують розчини пероксиду водню та каталізатора так, щоб їх суміш повністю опинилась в одному з її колін. Після насичення розчину киснем, який утворюється внаслідок розкладу Н2О2, газ почне виділятися з пробірки, і рівень води в газовій бюретці знижуватиметься.
З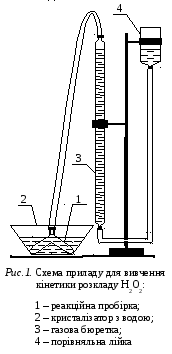 вільняють
зі штативу порівняльну лійку і, тримаючи
її поряд з газовою бюреткою, поступово
опускають услід за рівнем води в бюретці.
Щоб тиск газу в бюретці у будь-який
момент часу відповідав зовнішньому,
рівні води в бюретці та порівняльній
лійці повинні бути однаковими.
вільняють
зі штативу порівняльну лійку і, тримаючи
її поряд з газовою бюреткою, поступово
опускають услід за рівнем води в бюретці.
Щоб тиск газу в бюретці у будь-який
момент часу відповідав зовнішньому,
рівні води в бюретці та порівняльній
лійці повинні бути однаковими.
Секундомір вмикають тоді, коли рівень води в бюретці досягне позначки "0". Далі записують поточний час від початку реакції (пуску секундоміра), що відповідає виділенню об’єму кисню, який кожного наступного разу збільшується на 3 см3.
Отримавши таким чином 12 – 14 значень загального об’єму газу для відповідних проміжків часу, порівняльну лійку знов закріплюють на штативі так, щоб рівень води в ній був значно нижчий за рівень води у бюретці, і чекають повного розкладу пероксиду. Реакція вважається закінченою тоді, коли рівень води в газовій бюретці вже не змінюється.
Після
завершення реакції для визначення
кінцевого (максимального) об’єму газу,
що виділився, порівняльну лійку розміщують
поруч з бюреткою так, щоб вода в них була
на одному рівні, і вимірюють об’єм О2
в бюретці (![]() ).
Експериментальні дані записують у табл.
1.1 (див. звіт).
).
Експериментальні дані записують у табл.
1.1 (див. звіт).
За
даними табл. 1.1 будують графік
![]() .
Оскільки зазначена залежність не є
прямолінійною, то нульовий порядок, для
якого притаманна така зміна концентрації
продукту з часом, виключається. Тому за
методом підстановки перевіряють перший
порядок реакції, використовуючи рівняння
(1.5). Враховуючи те, що у формулі (1.5)
початкова концентрація пероксиду водню
.
Оскільки зазначена залежність не є
прямолінійною, то нульовий порядок, для
якого притаманна така зміна концентрації
продукту з часом, виключається. Тому за
методом підстановки перевіряють перший
порядок реакції, використовуючи рівняння
(1.5). Враховуючи те, що у формулі (1.5)
початкова концентрація пероксиду водню![]() є пропорційною
є пропорційною![]() ,
ах –
об’єму кисню
,
ах –
об’єму кисню
![]() ,
який виділився за певний проміжок часу,
константи швидкості обчислюють за
рівнянням:
,
який виділився за певний проміжок часу,
константи швидкості обчислюють за
рівнянням:
![]() . (1.15)
. (1.15)
Константи
швидкості розраховують для 5 – 6 точок
середньої частини кінетичної кривої
![]() (за вказівкою викладача). Сталість
одержаної за рівнянням (1.15) константи
швидкості реакції вказує на те, що
розклад пероксиду водню перебігає за
першим порядком. Обчислюють середнє
значення
(за вказівкою викладача). Сталість
одержаної за рівнянням (1.15) константи
швидкості реакції вказує на те, що
розклад пероксиду водню перебігає за
першим порядком. Обчислюють середнє
значення![]() .
.
Середнє
значення константи швидкості можна
визначити також графічно як тангенс
кута нахилу прямої в координатах
![]() .
.
Звіт
Дані експерименту:
![]() 4
см3;
4
см3; ![]() см3;
см3; ![]() моль/дм3.
моль/дм3.
Таблиця 1.1. Залежність об’єму кисню, що виділився внаслідок перебігу реакції, від часу
|
|
0 |
3 |
6 |
9 |
12 |
15 |
18 |
21 |
24 |
27 |
30 |
33 |
36 |
|
|
0 |
|
|
|
|
|
|
|
|
|
|
|
|
![]() см3.
см3.
Обчислення концентрації КІ у реакційній суміші:
 моль/дм3.
моль/дм3.Побудова за даними табл. 1.1 графіка
 .
.Розрахунки для табл. 1.2:
Таблиця 1.2. Дані для визначення константи швидкості та порядку реакції
|
№ виміру |
Час
з початку реакції
|
Об’єм
газу
|
|
k, с -1 |
|
1 6 |
|
|
|
|
![]() с
-1
с
-1
Побудова за даними табл. 1.2. графіка
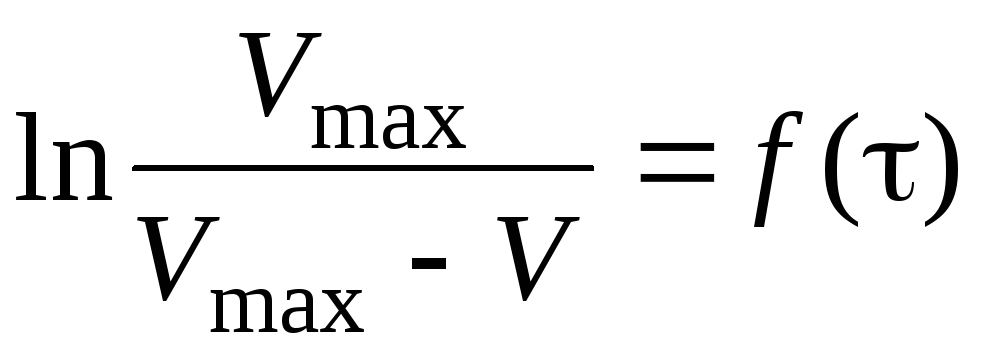 та обчислення
та обчислення
за
ним
![]() с
-1.
с
-1.
Кінетичне рівняння реакції:
Визначення часу напівперетворення:
а)
за формулою
![]() с;
с;
б)
за графіком
![]() :
:![]() с.
с.
Побудова графіка
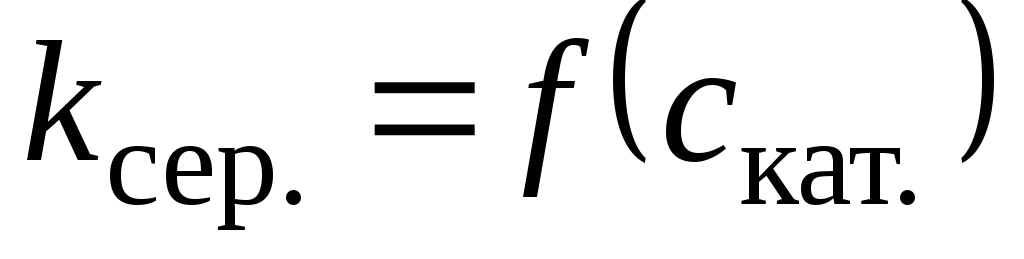 за даними табл. 1.3 та визначення константи
швидкості реакції без каталізатора:
за даними табл. 1.3 та визначення константи
швидкості реакції без каталізатора: с
-1.
с
-1.
Таблиця 1.3. Залежність константи швидкості реакції від концентрації каталізатора
-
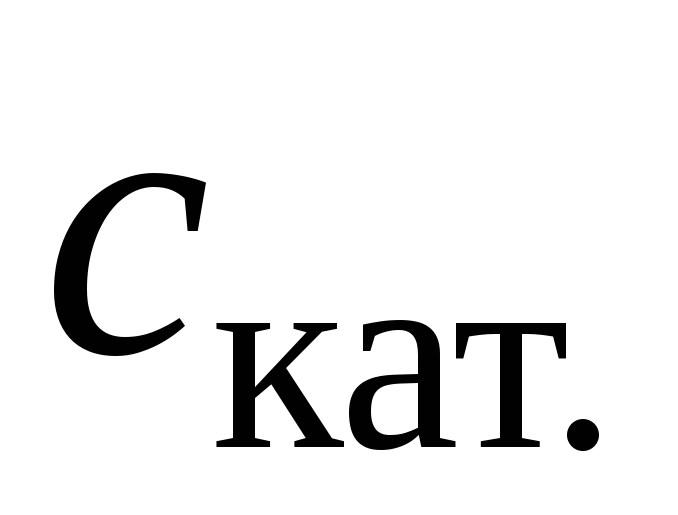 ,
моль/дм3
,
моль/дм3 ,
с
-1
,
с
-1
Розрахунок питомої активності каталізатора:
![]()
![]() .
.
Висновок.
ЛІТЕРАТУРА: [1, с. 523 – 527, 533 – 542, 616 – 623; 2, с. 290 – 293, 297 – 310, 427 – 433; 3, с. 318 – 329, 401 – 406; 4, с. 322 – 338, 344, 365 – 367; 5, с. 195 – 201, 204 – 205; 6, с. 217 – 225, 229 – 233; 8, с. 43 – 46].
Робота 1.2. Дослідження впливу температури на швидкість процесу розкладу пероксиду водню у водному розчині.
Мета роботи: визначити залежність константи швидкості реакції розкладу пероксиду водню у водному розчині від температури і обчислити енергію активації реакції.
Порядок виконання роботи наведено в роботі 1.1, але визначення проводять за різних температур і з однаковою концентрацією каталізатора КІ.
Звіт
Дані експерименту:
температура
досліду
![]() о
С;
о
С;
![]() см3;
см3;
![]() см3.
см3.
Таблиця 1.4. Залежність об’єму кисню, що виділився наслідок перебігу реакції, від часу
|
|
0 |
3 |
6 |
9 |
12 |
15 |
18 |
21 |
24 |
27 |
30 |
33 |
36 |
|
|
0 |
|
|
|
|
|
|
|
|
|
|
|
|
![]() см3.
см3.
Побудова за даними табл. 1.4 графіка
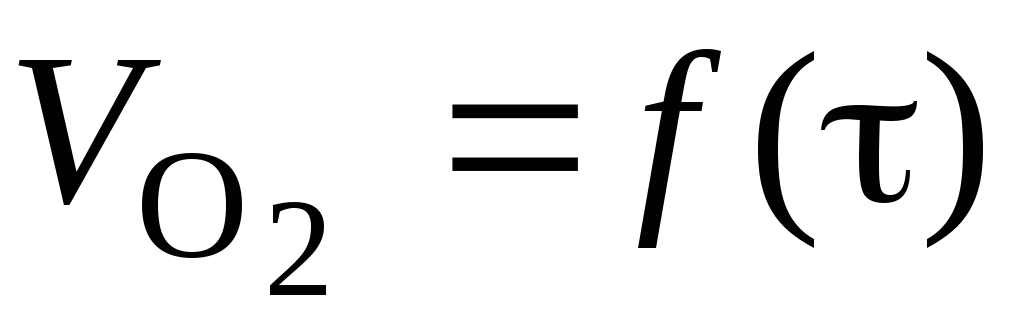 .
.Розрахунки для табл. 1.5:
Таблиця 1.5. Дані для визначення константи швидкості та порядку реакції
|
№ виміру |
Час
з початку реакції
|
Об’єм
газу
|
|
k, с -1 |
|
1 . . 6 |
|
|
|
|
![]() с
-1
с
-1
Кінетичне рівняння реакції:
Дані для графічного визначення енергії активації реакції:
Таблиця 1.6. Залежність константи швидкості реакції від температури
-
t, o C
T, K
 ,
К-1
,
К-1 ,
см3
,
см3 ,
с
-1
,
с
-1
t в лабораторії
25
32
39
Побудова графіка
 та визначення на його основі за формулою
(1.13) енергії активації реакції.
та визначення на його основі за формулою
(1.13) енергії активації реакції.Висновок.
ЛІТЕРАТУРА: [1, с. 528 – 531; 2, с. 313 – 315; 3, с. 337 – 341; 4, с. 338 – 346; 5, с. 202 – 205; 6, с. 235 – 236; 7, с. 217; 8, с. 46 – 47].
Робота 1.3. Дослідження кінетики реакції окиснення йодиду калію персульфатом калію.
Мета роботи: визначити частинний порядок реакції за персульфатом калію, установити залежність константи швидкості реакції від температури і обчислити енергію активації.
Досліджувана реакція перебігає за рівнянням:
К2S2O8 + 2KI = 2K2SO4 + I2. (1.16)
Якщо йодид калію взято у великому надлишку, то його концентрація в ході реакції практично не змінюється. В цьому випадку реакція має порядок, що відповідає частинному порядку за персульфатом калію.
В одну склянку або колбу мірною піпеткою наливають 50 см3 0,04–нормального розчину К2S2O8, а в другу – 50 см3 0,4–нормального розчину KI. Склянки занурюють у налаштований на певну температуру термостат і витримують приблизно 7 – 10 хвилин для нагрівання реагентів до температури досліду. Після цього розчини зливають, залишаючи ємність з досліджуваною системою в термостаті, та вмикають секундомір. Реакційна суміш повинна бути закритою, тому що йод легко випаровується.
Через певні проміжки часу (за вказівкою викладача, залежно від температури досліду) відбирають піпеткою 10 см3 суміші. За температур 32 о С і 39 о C проби бажано відбирати кожні 4 хвилини; за 25 о C – кожні 5 хвилин; за кімнатної температури – залежно від її значення, враховуючи те, що сумарна кількість титрувань складає 9. Пробу виливають у конічну колбу, яка містить приблизно 100 см3 холодної дистильованої води. В результаті виливання у воду концентрація розчину зменшується і він охолоджується, внаслідок чого реакція значно сповільнюється. Тому часом відбору проби слід вважати момент виливання її у воду.
Йод, який виділився під час реакції (1.16), титрують 0,01–нормальним розчином тіосульфату натрію за наявності крохмалю до зникнення синього забарвлення. Реакція, що перебігає при титруванні, має вигляд:
I2 + 2Na2S2O3 = 2NaI + Na2S4O6.
Експеримент припиняють тоді, коли витрачений на титрування трьох послідовних проб об’єм Na2S2O3 залишається практично незмінним.
Будують
графік
![]() та розраховують константи швидкості
реакції
(якщо
внаслідок похибок експерименту окремі
точки лежать поза плавною кривою, то ці
дані не використовують для розрахунку
константи швидкості). Припускаючи, що
частинний порядок за К2S2O8
є
першим, константу швидкості реакції
обчислюють за рівнянням (1.5). До формули
(1.5) входить концентрація вихідної
речовини (персульфату калію), а титруванням
визначають концентрацію продукту
реакції (І2).
Кількість йоду, що виділяється за певний
проміжок часу з початку реакції,
пропорційна кількості персульфату,
який реагує за цей час, і відповідає
об’єму Na2S2O3,
що витрачається на титрування реакційної
проби. Кінцева концентрація йоду
пропорційна початковій концентрації
К2S2O8
і, відповідно, максимальному об’єму
Na2S2O3.
Тому замість х
у формулу (1.5) підставляють об’єм
тіосульфату V,
який витрачено на титрування проби в
певний момент часу, а замість
та розраховують константи швидкості
реакції
(якщо
внаслідок похибок експерименту окремі
точки лежать поза плавною кривою, то ці
дані не використовують для розрахунку
константи швидкості). Припускаючи, що
частинний порядок за К2S2O8
є
першим, константу швидкості реакції
обчислюють за рівнянням (1.5). До формули
(1.5) входить концентрація вихідної
речовини (персульфату калію), а титруванням
визначають концентрацію продукту
реакції (І2).
Кількість йоду, що виділяється за певний
проміжок часу з початку реакції,
пропорційна кількості персульфату,
який реагує за цей час, і відповідає
об’єму Na2S2O3,
що витрачається на титрування реакційної
проби. Кінцева концентрація йоду
пропорційна початковій концентрації
К2S2O8
і, відповідно, максимальному об’єму
Na2S2O3.
Тому замість х
у формулу (1.5) підставляють об’єм
тіосульфату V,
який витрачено на титрування проби в
певний момент часу, а замість
![]() – максимальний об’єм тіосульфату
– максимальний об’єм тіосульфату![]() ,
що відповідає закінченню реакції.
,
що відповідає закінченню реакції.
Результати експерименту записують у табл. 1.7 (див. звіт).
Для визначення енергії активації реакції аналогічний дослід проводять
за 3 – 4 температур. Отримані результати для всіх температур зводять у
табл.
1.8 (див. звіт). За даними табл. 1.8
будують графік
![]() ,
,
визначають тангенс кута нахилу прямої і за рівнянням (1.13) обчислюють енергію активації реакції.
Звіт
Дані експерименту:
температура
досліду
![]() о
C.
о
C.
Таблиця 1.7. Залежність об’єму Na2S2O3, який витрачено на титрування реакційної проби, від часу
|
Час
від початку реакції
|
0 |
|
|
|
|
|
|
|
|
|
|
Об’єм
Na2S2O3
|
0 |
|
|
|
|
|
|
|
|
|
![]() см3.
см3.
Побудова за даними табл. 1.7 графіка
 .
.Обчислення за формулою
 констант швидкості реакції
констант швидкості реакції
(хв.-1):
![]() ;
;
![]() ;
;![]() ; ……..
; ……..![]() ;
;![]()
Висновок стосовно частинного порядку реакції за К2S2O8 та її кінетичне рівняння:
Дані для графічного визначення енергії активації досліджуваної реакції:
Таблиця 1.8. Залежність константи швидкості реакції від температури
|
t, о C |
|
|
|
|
|
|
t в лабораторії |
|
|
|
|
|
|
25 |
|
|
|
|
|
|
32 |
|
|
|
|
|
|
39 |
|
|
|
|
|
Побудова графіка
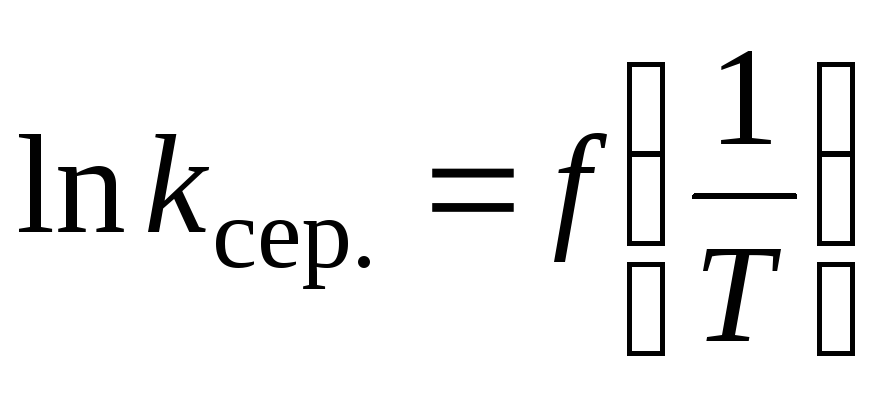 та визначення за ним енергії активації
реакції на основі формули (1.13).
та визначення за ним енергії активації
реакції на основі формули (1.13).
Висновок.
ЛІТЕРАТУРА: [1, с. 523 – 542, 616 – 623; 2, с. 290 – 310, 313 – 315, 427 – 433; 3, с. 318 – 329, 337 – 341, 402 – 406; 4, с. 322 – 346; 5, с. 195 – 206; 6, с. 217 – 225, 229 – 236; 8, с. 43 – 47].
Робота 1.4. Дослідження кінетики реакції окиснення йодиду калію персульфатом калію.
Мета роботи: визначити частинний порядок реакції за йодидом калію, дослідити залежність константи швидкості реакції від температури, обчислити енергію активації і установити загальний порядок процесу.
Досліджувана реакція перебігає за рівнянням (1.16).
З метою визначення частинного порядку реакції за KI у надлишку беруть К2S2O8. В одну склянку або колбу мірною піпеткою наливають 50 см3 0,4–нормального розчину персульфату калію, а в другу – 50 см3 0,04–нормального розчину йодиду калію. Склянки занурюють у термостат, який налаштовано на певну температуру. Далі робота виконується так само, як і попередня 1.3.
Звіт
Пункти 1 – 6 оформлюються так само, як у роботі 1.3.
Обчислення загальної константи швидкості, написання загального кінетичного рівняння реакції, визначення загального порядку реакції.
ЛІТЕРАТУРА: [1, с. 523 – 542, 616 – 623; 2, с. 290 – 310, 313 – 315, 427 – 433; 3, с. 318 – 329, 337 – 341, 402 – 406; 4, с. 322 – 346; 5, с. 195 – 206; 6, с. 217 – 225, 229 – 236; 8, с. 43 – 47].