

22.4. Теории хроматографического разделения
Рис.
22.3.Хроматограмма,
полученная при ГЖХ-определении аминазина
в печени (внутренний стандарт - бромгексин)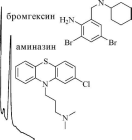
сорбции для молекул различных веществ различна, то после повторения большого числа элементарных актов хроматографического разделения при прохождении смеси веществ через слой сорбента происходит разделение её на отдельные компоненты. Положение и вид хрома- тографических зон разделяемых веществ зависят от формы изотермы сорбции, скорости установления равновесия, степени диффузии вещества в подвижной фазе.
Изотермой сорбцииназывается зависимость концентрации вещества, сорбированного неподвижной фазой, от его концентрации в подвижной фазе при постоянной температуре. Если изотерма сорбции линейна, установление равновесия происходит мгновенно и степень диффузии вещества в подвижной фазе пренебрежимо мала, идеальный хроматографический пик описывается кривой нормального распределения.
Для объяснения причин размывания хроматографических зон используются две теории: теоретических тарелок и кинетическая теория.
Теория теоретических тарелок предполагает, что:
каждая хроматографическая колонка состоит из некоторого количества одинаковых по величине абстрактных узких слоёв, называемых теоретическими тарелками, на каждой тарелке происходит один элементарный акт сорбции-десорбции;
на каждой тарелке происходит мгновенное установление равновесия между веществом, находящимся в подвижной и неподвижной фазе;
переход вещества с одной тарелки на другую происходит дискретно - при попадании на тарелку новой порции элюента равновесие нарушается, и часть вещества мгновенно переносится на следующую тарелку, где вновь мгновенно наступает равновесие и т. д.;
на любой тарелке в любой момент времени число сорбируемых частиц вещества значительно больше числа сорбируемых частиц растворителя, изотерма сорбции является линейной.
Количественной характеристикой хроматографической колонки являются: высота эквивалентная теоретической тарелке(H) и число теоретических тарелок(N).
н=L
N
N=L
H
Высота эквивалентная теоретической тарелке представляет собой дисперсию, приходящуюся на единицу длины колонки. Чем
меньше H и большеN, тем в меньшей степени происходит размывание пика и тем эффективнее хроматографическое разделение (рис. 22.4).

Рис.
22.4.Хроматограммы
вещества X, полученные на колонках с
различной эффективностью
(размерность оси абсцисс одинакова)
(t
>
2
(t
>tR
2
N
=
=
kx
1
а
I
Wx
J
где
tR
-
время удерживания,
kx
-
коэффициент, величина которого зависит
от того, на каком уровне измеряется
ширина пика
wx.
Если пик представляет собой кривую нормального распределения, то ширина пика у основания равна 4а, на половине высоты - 2,35а, на 60,7% высоты (между точками перегиба) - 2а (рис. 22.5) и
т.д. Приизмерении ширины пика у основания коэффициентkx бу-
2 2 дет равен 16 (4 ), на половине высоты - 5,54 (2,35 ) и т.д.
h
0,607h
0,5h
Рис.
22.5.Свойства
идеального хроматографического пика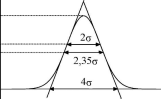
Число теоретических тарелок является мерой эффективности колонки и обычно постоянно для всех пиков на хроматограмме. Так как N является постоянной величиной, то при увеличении времени удерживания ширина пика увеличивается.
Число
теоретических тарелок можно рассчитать:
Согласно кинетической теории хроматографииразмывание хроматографических пиков обусловлено одновременным действием трёх независимых друг от друга процессов:
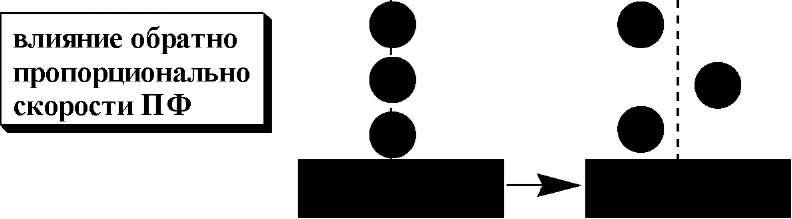
продольная
диффузия ]
B
РАЗМЫВАНИЕ ХРОМАТОГРАФИЧЕСКОГО ПИКА
сопротивление массопереносу^ C
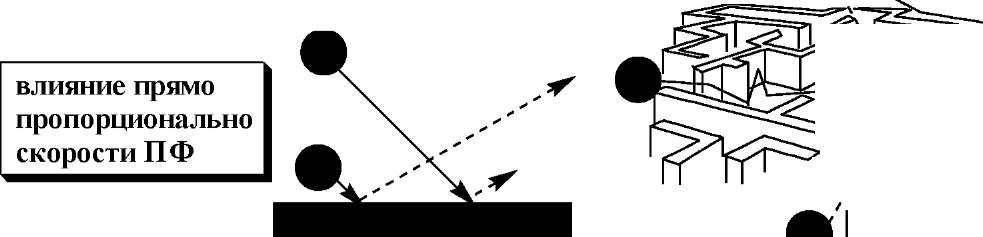
В
насадочной колонке молекулы компонентов
разделяемой пробы движутся между
частицами сорбента по разным траекториям.
Л
вихревая диффузия ] A
|
|
влияние |
|
/ / 1 * -——_ |
не зависит |
|
|
от скорости |
|
/ / / / ч/ |
ПФ |
Суммарное влияние вихревой диффузии, продольной диффузии и сопротивления массопереносу на величину высоты эквивалентной теоретической тарелке описывается уравнением Ван-Деемтера.
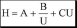
где U - линейная скорость подвижной фазы
H
CU
B/U
опт U
Рис.
22.6.ЗависимостьH от линейной скорости
газа-носителя
H
U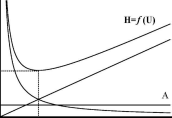
Зависимость H отU для газовой хроматографии (насадочная колонка) показана на рис. 22.6. Оптимальную скорость газа-носителя, при которой величина Н минимальна, можно рассчитать по формуле:
|
|
B |
|
U опт ^ |
C |
В жидкостной хроматографии величина B практически не вносит вклад в размывание хроматографического пика (вязкость жидкости значительно больше вязкости газа), поэтому зависимость Н отUвыглядит по-другому (как?).
Для характеристики эффективности разделения компонентовсмеси, используют коэффициент разделения (а) и разрешение(RS).
Если
a = 1, то разделение
невозможно. Разрешение
рассчитывается по следующей формуле:


Разделение двух пиков считается полным, если RS > 1,5(приRS = 1,5 расстояние между максимумами пиков составляет 6а, степень перекрывания пиков 0,13%) - рис. 22.6.
Rs=_L6l=1,5S 4а + 4а
Рис.
22.6.Перекрывание
пиков при различной величинеRS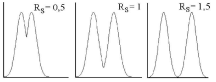
Число теоретических тарелок, необходимое для разделения с заданным разрешением, равно:
|
N = 16R2 |
f k 2+1 1 |
2 |
f a 1 |
2 |
|
I k 2J |
|
la-1J |
|