

3.3. Методы определения общего содержания азота почвы.
Азот относится к числу важнейших элементов питания растений, поэтому общий запас азота в почве является одним из показателей ее потенциального плодородия. Общее содержание азота варьирует от 0,03-0,05% в песчаных по гранулометрическому составу почвах, до 0,6-0,8% в тучных глинистых черноземах.
В почве азот находится преимущественно в органическом веществе. В гумусовых горизонтах почв на долю азота органических соединений приходится 93 – 97 % от его общего содержания, при этом до 70 – 90 % азота входит в состав специфических гумусовых веществ (табл. 22).
Таблица 22. Распределение запасов азота по группам гумусовых веществ (по данным Д.С. Орлова, 1992)
|
Почва |
Всего азота, кг/га |
Азот гуминовых кислот |
Азот фульвокислот |
Азот гумина |
Азот декальцината и неспецифических веществ | ||||
|
кг/га |
в % от общего |
кг/га |
в % от общего |
кг/га |
в % от общего |
кг/га |
в % от общего | ||
|
Дерново-среднеподзолистая |
3560 |
341 |
9,6 |
620 |
17,4 |
1628 |
45,7 |
625 |
17,6 |
|
Типичный чернозем |
9890 |
1720 |
17,4 |
864 |
8,7 |
5158 |
52,1 |
1224 |
12,4 |
|
Серозем |
3420 |
348 |
10,2 |
326 |
9,5 |
1824 |
53,3 |
555 |
16,2 |
Поскольку преобладающая часть азота почвы связана с органическим веществом, то его содержание и запасы, а так же распределение по почвенному профилю в основном повторяют закономерности профильного накопления и распределения органических веществ.
Для определения общего содержания азота в почве, чаще всего используют либо газоволюмометрические методы, основанные на термическом разложении органического вещества и измерении объема выделившегося азота, либо методы, предусматривающие мокрое озоление органического вещества. Среди последних методов наиболее широкое применение нашли методы Кьельдаля и Тюрина. Определение азота этими методами включает три этапа: озоление органического вещества, отгонку аммиака, количественное определение азота.
3.3.1. Определение общего содержания азота методом Кьельдаля.
Метод основан на разложении (озолении) органического вещества почвы концентрированной серной кислотой при кипячении и последующем количественном учете образующегося азота.
При взаимодействии органического вещества с кипящей серной кислотой углерод и водород органических соединений окисляются до CO2 и H2O, а окислению азота препятствует образующийся в процессе реакции диоксид серы, поэтому он остается в восстановленной форме и превращается в сульфат аммония. Схематично, на примере аланина, являющегося одним из компонентов гумуса почвы, реакция будет протекать следующим образом:
2CH3CHNH2COOH + 13H2SO4 (NH4)2SO4 + 6CO2 + 16H2O + 12SO2
Раствор, содержащий сульфат аммония переносят в перегонную колбу, в которой аммиак вытесняется из сернокислой соли щелочью:
(NH4)2SO4 + 2NaOH 2NH3 + 2H2O + Na2SO4
Выделяющийся аммиак отгоняют в приемник, где он улавливается определенным объемом титрованного раствора серной кислоты:
2NH3 + H2SO4 (NH4)2SO4
По оставшемуся свободным и определенному титрованием раствором щелочи, количеству серной кислоты, находят количество поглощенного аммиака.
Для ускорения сжигания органического вещества прибавляют катализаторы – медь, ртуть или селен, а для повышения температуры кипения – сернокислый калий. Чтобы диоксид серы не улетучивался, колбу закрывают особой стеклянной пробкой с шарообразным расширением на одном конце и суженной на другом.
Метод Кьельдаля достаточно точен и широко применяется в исследованиях. Однако нитратный азот этим методом не определяется, и при его высоком содержании в почве можно получить заниженные результаты.
В этом случае рекомендуется метод Иодльбауэра, отличающийся от метода Кьельдаля тем, что почву обрабатывают не серной, а фенолсерной кислотой. Фенолсерную кислоту приготавливают растворением 40 г чистого фенола в 1 л концентрированной H2SO4.
Ход анализа. Из образца почвы, приготовленного для определения общего гумуса, в сухую чистую пробирку отвешивают на аналитических весах 1-5 г с таким расчетом, чтобы в ней содержалось 20-40 мг азота. На запаянный конец пробирки надевают отрезок резиновой трубки и с его помощью вводят пробирку в сухую колбу Кьельдаля. Пробирку опускают как можно глубже, почти до дна колбы, и осторожно высыпают почву так, чтобы она не распылилась.
Пробирку вынимают из каучуковой трубки и взвешивают. По разности между весом пробирки с почвой и без нее находят величину взятой навески почвы для определения содержания общего азота.
Сжигание. В колбу Кьельдаля со взятой навеской почвы добавляют две-три крупинки селена или 1 г сернокислой меди (или 0,5 г окиси меди) и 10 мг K2SO4. По стенкам, стараясь смыть пылинки и смочить всю почву, приливают 10 мл концентрированной H2SO4. Содержимое колбы осторожно перемешивают круговыми движениями, стараясь не размазать суспензию по грушевидному расширению колбы. Пока вся почва не будет смочена кислотой, нагревание содержимого начинать нельзя, поскольку сухая почва при нагревании пригорает и происходит частичная потеря азота. Кроме того, в том месте, где почва не смочена кислотой, колба может треснуть от перегрева. Колбу закрывают свободнолежащей стеклянной пробкой, ставят для озоления в вытяжном шкафу на нагревательный прибор в наклонном положении, чтобы жидкость при кипении не выбрасывало из колбы, и начинают медленное нагревание. Нагревать начинают на слабом огне. Когда начнется обильное выделение белого «дымка», нагрев увеличивают и содержимое колбы доводят до слабого кипения, которое продолжается, пока раствор не станет прозрачным и почти бесцветным, а осадок белым. Кипение серной кислоты все время должно быть слабым, во избежание потерь (NH4)2SO4. В процессе кипения не должно происходить вспучивание смеси. В случае сильного вспенивания, колбу снимают с огня, дают ей немного охладиться и перемешивают содержимое, следя, чтобы пена не поднималась выше ее грушевидного расширения, затем снова ставят колбу на огонь.
После просветления жидкости и побеления осадка почвы смесь кипятят еще в течение 1-2 часов для полной уверенности в окончании реакции.
Отгонка аммиака. Для отгонки собирают прибор, состоящий из термостойкой плоскодонной отгонной колбы на 750 или 1000 мл, каплеуловителя, холодильника и аллонжа, опущенного в приемную коническую колбу на 250 мл (рис. 2).
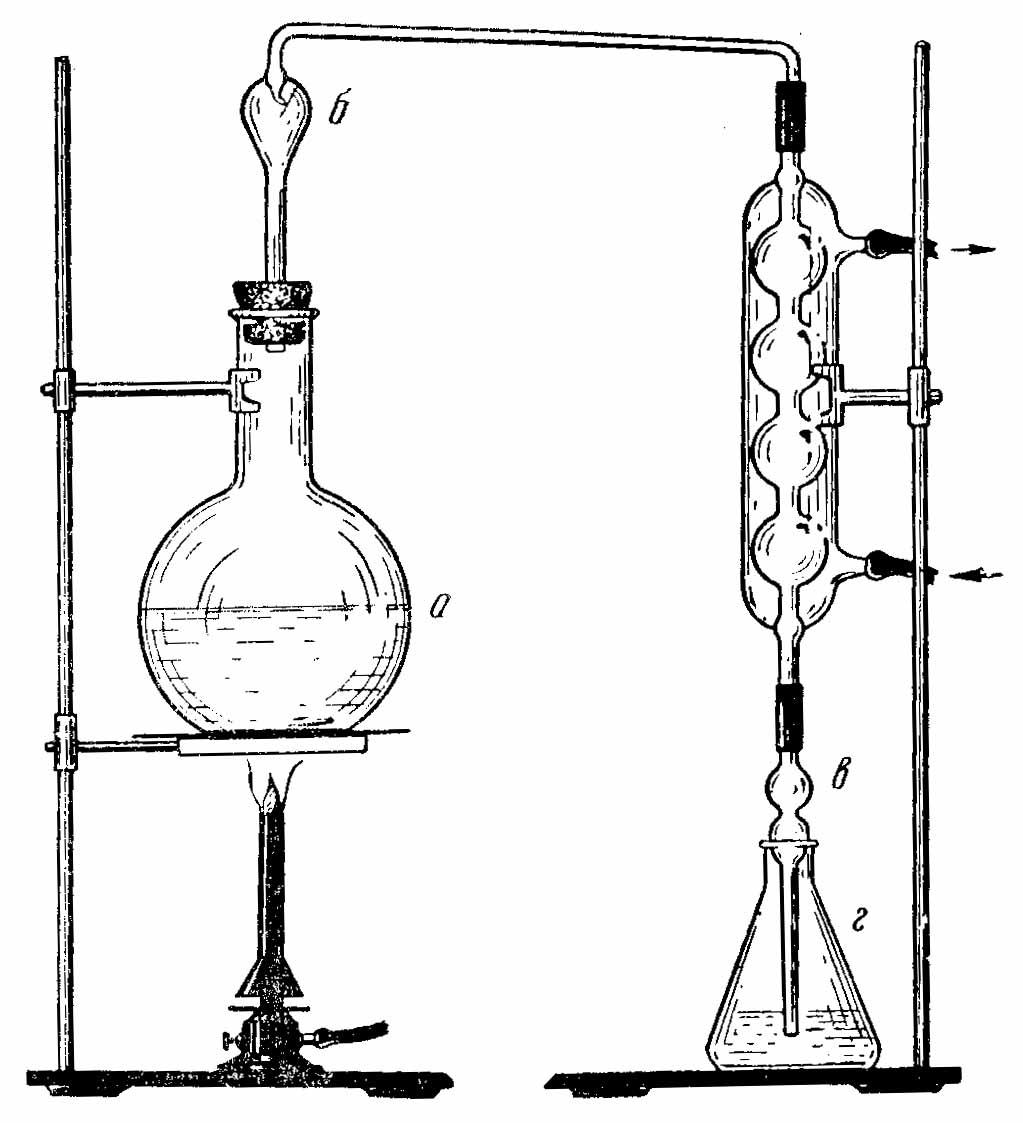
Рис. 4. Прибор для отгонки аммиака: а– перегонная колба,б– каплеуловитель,в– дистилляционная трубка,г– приемник с титрованным раствором кислоты
В охлажденную колбу Кьельдаля, поставленную наклонно, осторожно, небольшими порциями, обмывая стенки и пробку, приливают дистиллированную воду до объема около 50 мл и переносят раствор в отгонную колбу. Осадок нужно тщательно отмыть многократными небольшими порциями дистиллированной воды, сливая промывные воды в отгонную колбу. Всего в отгонной колбе должно быть не более 300 – 400 мл жидкости. Переносить остаток отмытой почвы не обязательно.
Вслед за этим подготавливают приемник. В коническую колбу на 250 мл приливают из бюретки 20 мл 0,02-0,05-0,1 н раствора H2SO4, в зависимости от содержания азота, и добавляют несколько капель индикатора Гроака, окрашивающего раствор кислоты в красно-фиолетовый цвет. Эту колбу с титрованной кислотой ставят под аллонж на подставку так, чтобы его конец был слегка погружен в кислоту примерно на 5 мм.
В отгонную колбу с перенесенным раствором по стенке в наклонном положении осторожно приливают 40 %-ный раствор NaOH в 4-кратном объеме к взятой для сжигания кислоте. Спускаясь по стенкам колбы, щелочь стекает на дно, не смешиваясь с более легкой жидкостью, что уменьшает опасность потери аммиака. Прибавляют 2 – 3 капли фенолфталеина и маленький кусочек гранулированного цинка или стеклянные капилляры, чтобы обеспечить равномерное кипение жидкости. Колбу присоединяют к перегонному аппарату, плотно закрывая смоченной для герметичности дистиллированной водой резиновой пробкой с каплеуловителем. Для обеспечения полной герметичности прибора пробку около трубки каплеуловителя и в месте соприкосновения с расширением горла колбы смачивают несколькими каплями дистиллированной воды, следя за тем, чтобы вода не стекала по колбе и не смачивала ее дно. Пускают воду в холодильник, а содержимое колбы тщательно перемешивают круговыми движениями. Малиновая окраска жидкости в колбе свидетельствует о щелочной реакции раствора. Недостаточно полное перемешивание может привести к локальному перегреву жидкости на дне перегонной колбы, бурному вскипанию и даже взрыву колбы. Сразу же после перемешивания колбу устанавливают на асбестовой сетке и горло ее слегка закрепляют. Раствор постепенно нагревают до кипения, которое должно быть равномерным и не сильным до конца отгонки.
В процессе отгонки аммиака происходит засасывание жидкости из колбы-приемника в трубку с шарообразными расширениями. Перебрасывание приемной жидкости в отгонную колбу сопровождается опасным взрывом. Во избежание взрыва при интенсивном, непрекращающемся засасывании жидкости в трубку, приемную колбу нужно опустить и вынуть трубку из раствора. Если это пришлось сделать до начала перегонки воды, то анализ необходимо повторить из-за возможных потерь NH3.
Через некоторое время после начала кипения (6-8 минут), когда будет происходить отгонка воды, приемную коническую колбу необходимо опустить и трубку вынуть из раствора во избежание перебрасывания жидкости из приемника в отгонную колбу. Вода с растворенным аммиаком будет стекать в приемник. Через 30-40 минут проверяют полноту отгонки по лакмусовой бумажке. Если очередная капля отгона не вызывает посинения лакмусовой бумажки, то прекращают нагревание раствора и выключают холодильник. Конец трубки обмывают изнутри и снаружи дистиллированной водой, собирая ее в колбу-приемник. Избыток раствора серной кислоты в приемнике титруют раствором NaOH той же концентрации, что и кислота, взятая для поглощения аммиака. По количеству связанной аммиаком серной кислоты вычисляют содержание азота:
![]()
где V1 – количество серной кислоты, взятой в приемник, мл; V2 – количество щелочи, пошедшее на обратное титрование, мл; н1 – нормальность кислоты; н2 – нормальность щелочи; 0,014 – количество азота в граммах, соответствующее 1 мг-экв. аммиака; m – навеска абсолютно сухой почвы; 100 – коэффициент пересчета на 100 г почвы.
Пример расчета. В приемную колбу взято 20 мл 0,02 н. раствора H2SO4. На титрование остатка кислоты пошло 14,6 мл 0,022 н. раствора NaOH. Навеска почвы равна 1,016 г. Содержание азота составило:
![]()
Из полученной величины содержания азота в почве вычитают то количество азота, которое найдено при проведении холостого или контрольного опыта на чистоту реактивов.
Для анализа используют следующие реактивы:
1. Концентрированная серная кислота (пл. 1,84), не содержащая аммонийных солей.
2. 40 %-ный раствор NaOH. 400 г NaOH помещают в сухой фарфоровый стакан или термостойкую посуду с маркировкой на уровне 1 л. Приливают 600 мл дистиллированной воды, лишенной CO2 и тщательно перемешивают содержимое до полного растворения. При этом происходит сильный разогрев жидкости. Раствор закрывают бумагой и оставляют до следующего дня, после чего доводят до метки и переносят для хранения в сосуд, закрытый пробкой с хлоркальциевой трубкой, содержащей аскарит, поглощающий СО2.
3. Титрованный раствор NaOH и H2SO4. Растворы соответствующих концентраций готовят из фиксаналов.
4. Смешанный индикатор Гроака. 1 объем 0,4 % спиртового раствора метилового красного смешивают с 1 объемом 0,2 % спиртового раствора метиленового голубого. Раствор смешанного индикатора хранят в темной склянке.