

H-31_vms_lections (ВМС)
.pdfдействующая или периодически прилагаемая на определенный промежуток времени, а также циклическая. Зависимость деформации от температуры, получившая название термомеханической кривой и представленная в общем виде на рис. 9, позволяет сделать заключения о температурных интервалах существования полимера в том или ином физическом состоянии.
Рис. 9. Термомеханическая кривая линейного аморфного полимера.
Область I (рис. 9), в которой упругие деформации являются небольшими по величине и энергетическими по природе, соответствует стеклообразному состоянию. По достижении Тс начинает проявляться сегментальная подвижность и макромолекулы приобретают некоторую способность к конформационным переходам, хотя они еще и затруднены высокой вязкостью и осуществляются замедленно – температурный интервал Тс соответствует переходу полимера из стеклообразного в высокоэластическое состояние. При Тс достигает максимального значения угол сдвига фаз между напряжением и деформацией в случае воздействия циклических нагрузок и наблюдаются максимальные потери механической энергии. Температурная область II соответствует развитому высокоэластическому состоянию: фиксируемые в ней деформации являются равновесными высокоэластическими, обусловленными конформационными переходами макромолекул в поле механических сил. По мере приближения к интервалу текучести начинают постепенно проявляться взаимные перемещения макромолекул, т. е. развиваться деформация течения – выше ТТ полимер находится в вязкотекучем состоянии (область III).
Втех случаях, когда полимер имеет редкие поперечные связи, вязкотекучее состояние отсутствует, и высокоэластическая деформация проявляется вплоть до температуры начала разложения полимера, выше которой ход термомеханической кривой имеет сложный характер. При увеличении частоты пространственной сетки высокоэластическая деформация уменьшается и, когда число поперечных связей превысит одну на каждый сегмент, трехмерный полимер будет деформироваться как обычное твердое тело, т. е., высокоэластическая деформация перестанет проявляться.
Вряду высокомолекулярных линейных полимергомологов температура стеклования не зависит от молекулярной массы, т.к. она определяется лишь размером среднестатистического сегмента; в то же время с увеличением размеров цепей становятся более трудноосуществимыми их взаимные перемещения и они происходят при более высокой температуре. Следовательно, с ростом молекулярной массы линейного аморфного полимера происходит повышение температуры текучести, т. е. расширяется температурная область высокоэластического состояния.
Температура стеклования, определяемая термомеханическим методом, существенно зависит от времени действия силы на образец при каждом измерении деформации или от частоты при циклическом деформировании. Так с повышением частоты прилагаемой нагрузки температура стеклования повышается.
Простота и высокая информативность сделали термомеханический анализ весьма
http://www.mitht.org
популярным. По термомеханическим кривым устанавливают аморфность или кристалличность полимера, оценивают влияние пластификаторов и наполнителей, определяют критические температуры, такие как температуры размягчения, стеклования, высокоэластичности, плавления, кристаллизации.
Возможности термомеханического анализа могут быть использованы значительно шире и в том числе в технологическом плане. По термомеханической кривой возможна оценка влияния способа производства полимера на его свойства, корректировка температурных условий пластикации термопластов, оптимизация режимов формования изделий из расплава и из заготовок (пневмо- и вакуумформование), оценка влияния морфологии и свойств армирующих волокнистых наполнителей на термодеформационное поведение угле- и стеклопластиков.
5.Понятие о релаксации.
Вобщем виде релаксацией называется процесс перехода системы из неравновесного состояния в равновесное. Этот переход происходит вследствие теплового движения структурных элементов, из которых состоит данная система, и обусловлен, таким образом, термодинамическими причинами. Естественно, что все факторы, влияющие на подвижность элементов структуры рассматриваемой системы, влияют и на характер проявления ее релаксационных свойств. Так, повышение температуры, уменьшение энергии межмолекулярного взаимодействия и уменьшение размеров элементов структуры приводит к ускорению достижения системой равновесного состояния, т. е. к ускорению протекания релаксационных процессов.
Механические свойства системы меняются при протекании в ней релаксационных процессов. Если эти процессы происходят быстро по сравнению с временем наблюдения или временем эксплуатации изделия, то их можно и не учитывать. Если же скорость протекания релаксационных процессов сравнима со временем наблюдения или эксплуатации изделия, то совершенно необходимо знать закономерности релаксации, чтобы правильно прогнозировать изменение свойств материалов и изделий, а следовательно, и правильно направлять их свойства при создании этих материалов и изделий.
Для простой релаксирующей системы (в которой элементы структуры малы и энергия их взаимодействия друг с другом невелика) тепловое движение элементов структуры - величина, определяющая изменения в системе и скорость приближения системы к равновесию, пропорциональна величине изменения, прошедшего в системе. Если напряжение в образце Р, а скорость релаксации напряжения равна dР/dt, то скорость перехода к равновесному состоянию пропорциональна напряжению:
dР/dt= - Р/ ,
После разделения переменных и интегрирования (от 0 до и от Р0 до Р) получим:
Р = Р0е-t/
Это уравнение позволяет определить физический смысл . Как видно из уравнения, должно иметь размерность времени; пусть t = , тогда Р = Р0/е. Следовательно, - это
время, в течение которого начальное напряжение Р0 уменьшится в е раз. Величина называется временем релаксации системы. Различные системы характеризуются различными временами релаксации. В низкомолекулярных жидкостях с малой вязкостью времена релаксации составляют величины порядка 10-8 – 10-10 с. Полимерные макромолекулы вследствие очень больших размеров обладают малой подвижностью. Поэтому для макромолекул время релаксации может быть порядка суток и месяцев. Таким образом, если для низкомолекулярных жидкостей можно пренебречь протеканием релаксационных процессов вследствие их высокой скорости по сравнению с обычными скоростями приложения
http://www.mitht.org
внешних воздействий и наблюдения, то в полимерах надо всегда сопоставлять скорости внешних воздействий на образцы или изделия со скоростями внутренних изменений в них вследствие перестройки структуры, которая протекает в течение длительного времени.
Если, например, скорость деформирования образца или изделия из полимера меньше скорости протекания релаксационных процессов в нем, то последние успевают завершиться за время испытания или воздействия внешней силы. Если же скорость деформирования больше скорости релаксации, то равновесная деформация не достигается, и необходимо учитывать протекание релаксационных явлений, которые будут влиять на изменение формы образца или изделия из полимера с течением времени.
6. Зависимость релаксационных свойств полимеров от строения молекулярных цепей и характера их взаимодействия друг с другом.
Деформационные свойства полимеров обусловлены строением их молекулярных цепей и связаны с различными молекулярными механизмами их взаимодействия. Так, для аморфных полимеров характерны, например, следующие виды деформаций. Во-первых, гуковская упругость, обусловленная ограниченной подвижностью сегментов макромолекулярных цепей. Обычно считают, что этот вид деформации связан с растяжением валентных связей и углов, а потому величины деформации крайне малы, и материал ведет себя как стекло. Во-вторых, высокоэластичность, обусловленная свободой перемещения сегментов благодаря гибкости цепи и наличием флуктуационной сетки, препятствующей перемещению макромолекул в целом, т. е. процессу течения. В третьих, вязкое течение - скольжение макромолекул друг относительно друга, приводящее к необратимому деформированию материала. В реальном полимере деформация является вязкоупругой, т. е. она сочетает необратимую и обратимую деформации. Она связана с переходом полимерных цепей из их равновесных конформаций в неравновесные благодаря перемещению сегментов вследствие свободы вращения вокруг химических связей в цепи макромолекулы. Этот последний - четвертый вид деформации включает элементы вязкого течения, которые не являются истинным течением, а обратимы и могут исчезать со временем. Таким образом, вязкоупругость представляет собой как бы сочетание вязкого течения и высокой эластичности.
7. Релаксационные явления в высокоэластическом состоянии полимеров.
Рассмотрим протекание релаксационных процессов в аморфных полимерах в высокоэластическом состоянии.
В высокоэластическом состоянии наиболее полно реализуется подвижность сегментов макромолекул, причем взаимное расположение макромолекул при воздействии внешних усилий на полимер не изменяется. Таким образом, в этом состоянии можно наблюдать протекание релаксационных процессов, не осложненных течением макромолекул или гуковской деформацией. Деформации полимеров в высокоэластическом состоянии являются большими и обратимыми, а их обратимость, в отличие от гуковских деформаций протекает в течение длительного времени.
Ползучесть. Протекание релаксационных явлений в аморфных полимерах можно рассмотреть на примере ползучести. Ползучестью называется увеличение деформации полимера под действием постоянной нагрузки. Если к образцу полимера, взятому в виде прямоугольной полоски, подвесить постоянный груз Р (второй конец полоски закреплен неподвижно), то с течением времени будет происходить рост удлинения образца (рис. 10).
http://www.mitht.org
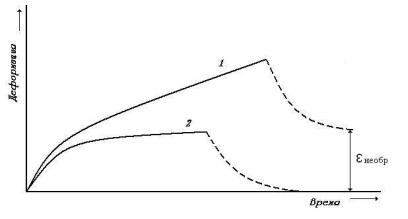
Рис. 10. Кривые ползучести полимеров:1 - несшитого и 2 – сшитого поперечными химическими связями; пунктир – уменьшение удлинения после снятия нагрузки.
Вследствие роста относительного удлинения такого образца ( = (l – l0)/ l0 его поперечное сечение уменьшается и напряжение непрерывно растет. Если испытывается полимер, не сшитый поперечными связями, то после быстрого роста удлинения в начале деформирования наступит период удлинения образца с постоянной скоростью, который может продолжаться неограниченно долго вплоть до разрыва образца (кривая 1). Если мы испытываем сшитый полимер, то после начального роста деформации ее величина стремится к некоторому пределу, тем большему, чем меньше поперечных связей в полимере и чем больше приложенный груз (кривая 2).
Кинетика развития ползучести имеет релаксационную природу и связана с проявлением вязкоупругих свойств аморфного полимера. В линейном несшитом образце под действием деформирующей силы происходит одновременное выпрямление первоначально свернутых макромолекул и их перемещение друг относительно друга вследствие вязкого течения. В этом случае развивается необратимая пластическая деформация. Постоянная скорость роста деформации такого образца связана с перемещением частично распрямленных макромолекул друг относительно друга. В сшитом образце поперечные связи препятствуют последнему процессу, и поэтому деформация достигает некоторого постоянного значения после перехода свернутых макромолекул в более выпрямленное и ориентированное по направлению действия силы состояние.
При этом деформирующая сила преодолевает внутри- и межмолекулярные взаимодействия макромолекул. Чем больше эти взаимодействия (наличие полярных групп в макромолекулах, например, полиамидов, полиэфиров и др.), тем труднее их преодолеть и тем медленнее развивается деформация в образце полимера. Наличие в макромолекулах боковых ответвлений (разветвленные структуры) затрудняет перемещение макромолекул или сегментов друг относительно друга и таким образом тоже увеличивает время развития деформации. Повышение температуры способствует ускорению движения сегментов и макромолекул и сокращает время развития деформации. Таким образом, факторы, уменьшающие время релаксации макромолекул способствуют увеличению скорости ползучести. Упругая гуковская деформация для аморфных полимеров в высокоэластическом состоянии мала и поэтому, ею можно пренебречь.
Если в какой-то момент времени снять нагрузку, действующую на образец, то поскольку в сшитом образце необратимые деформации вязкого течения отсутствуют, он полностью восстанавливает свою исходную форму: произойдет его быстрое сокращение вследствие скручивания макромолекул в результате теплового движения (рис. 10). Образец линейного (несшитого) полимера не возвращается в исходное состояние, так как необратимое перемещение макромолекул после снятия нагрузки не исчезает (так же как жидкость не восстанавливает свою форму при переливании ее из одного сосуда в другой). Однако и в линейном, и в сшитом полимере эти процессы релаксации протекают во времени. Перечисленные выше факторы, способствующие ускорению протекания релаксационных процессов, приводят к более быстрому восстановлению исходного состояния полимера.
http://www.mitht.org
Для ускорения протекания релаксационных процессов после снятия нагрузки образец можно подвергнуть нагреванию (не более 700С, чтобы не развились химические реакции с кислородом воздуха или термический распад химических связей). Так, например, растянутая при комнатной температуре до 400% удлинения пленка каучука сокращается после снятия нагрузки до 200-300%. Затем процесс релаксации резко замедляется и ускоряется только при нагревании образца до 500С.
Релаксация напряжения. Другим наглядным примером релаксации в полимере при изменении одного из параметров деформирования является изменение напряжения при сохранении постоянства деформации образца. Если быстро растянуть образец аморфного полимера до какой-то величины удлинения и закрепить его в этом положении, то можно проследить за изменением напряжения в образце с течением времени. С течением времени в таком образце наблюдается падение напряжения, так как после быстрого растяжения образца свернутые макромолекулы примут конформации, вытянутые в направлении растяжения. Однако флуктуационная сетка за короткий промежуток времени деформирования не успевает разрушиться. С течением времени тепловое движение стремится перевести макромолекулы в более вероятные для них свернутые конформации, и флуктуационная сетка, распадаясь под действием теплового движения сегментов макромолекул, создается вновь для более термодинамически вероятного состояния макромолекул. Естественно, что повышение температуры увеличивает интенсивность теплового движения элементов структуры и тем самым ускоряет протекание релаксационных процессов.
Если макромолекулы в образце не соединены друг с другом химическими поперечными связями, то последние не препятствуют их переходу в исходное, свернутое состояние, и после релаксации напряжение в образце упадет до нуля. Макромолекулы примут ту же форму, какая была до деформации. Если же образец содержит макромолекулы, связанные друг с другом химическими поперечными связями, которые не разрушаются в процессе релаксации, то они препятствуют восстановлению исходной формы макромолекулами. В такой системе напряжение упадет до некоторого равновесного значения, тем большего, чем больше поперечных связей содержится в структуре полимера.
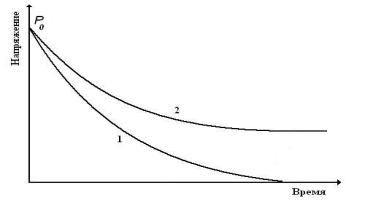
На рис. 11, показано протекание процесса релаксации напряжения в линейном и сшитом образцах полимера. В неполярных полимерах, где флуктуационная сетка, менее прочна, процессы релаксации протекают быстрее, чем в полярных полимерах.
Рис. 11. Релаксация напряжения в полимерах: 1 – несшитом, 2 – сшитом поперечными химическими связями.
8. Механические свойства полимеров в высокоэластическом состоянии.
Релаксационный характер деформации проявляется в том, что зависимость напряжения от деформации при нагружении и разгружении, как правило, не совпадают (рис. 12).
http://www.mitht.org
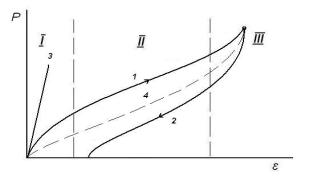
Рис. 12. Петля упругого гистерезиса: 1 – нагружение; 2 – разгружение; 3- быстрое нагружение – разгружение; 4 – равновесная кривая.
Если скорость деформации эластомера больше скорости перестройки формы макромолекул под действием растягивающего усилия, то деформация не успевает полностью развиться за время действия деформирующего усилия. Величина деформации образца будет ниже равновесной (рис. 12, 1), так как скорость перестройки формы макромолекул меньше скорости прилагаемого напряжения. Деформация частично обусловлена изменением расстояний между макромолекулами и их взаимным перемещением. На начальном участке кривой (I) деформация мала и имеет преимущественно гуковскую природу, на участке II происходит раскручивание макромолекул и преобладает высокоэластический механизм. На участке III деформируются частично выпрямленные макромолекулы, что связано с деформацией валентных углов, т. е. вновь преобладает гуковская деформация.
При разгрузке образца скорость разгружения больше скорости возвращения макромолекул в недеформированное состояние: образец не успевает полностью сократиться, и значение деформации оказывается в каждый момент времени разгружения больше равновесного. Таким образом, как видно из рис. 12, при неравновесной деформации кривые
нагрузка - разгрузка в координатах Р - не совпадают. Графически это выражается петлей гистерезиса. В итоге кривая разгрузки не попадает в начало координат, т. е. после полного снятия напряжения образец сохраняет некоторую остаточную деформацию, величина которой с течением времени уменьшается вследствие возвращения макромолекул в исходную свернутую конформацию. Если при деформации не происходило необратимое смещение макромолекул вследствие их вязкого течения, то по достижении макромолекулами равновесной свернутой конформации остаточное удлинение образца равно нулю. Если же вязкое течение имело место, то образец никогда уже не примет свою прежнюю форму, и истинная остаточная деформация в нем соответствует величине смещения макромолекул.
Гистерезисная петля на рис. 12 может и не проявиться, если возрастание нагрузки происходит очень быстро или, наоборот, очень медленно. При быстром нагружении в образце не успевают произойти необходимые для развития высокоэластической деформации перегруппировки сегментов и конформационные переходы, и образец полимера будет вести себя как обычное твердое тело – в нем обнаружится только практически мгновенно развивающаяся обычная упругая деформация (рис. 12, кривая 3).
При медленном осуществлении цикла нагружение - разгружение в образце успевают произойти необходимые конформационные переходы макромолекул, и фиксируемая деформация является равновесной высокоэластической.
Площадь, ограничиваемая гистерезисной петлей напряжение - деформация, пропорциональна работе, теряемой в одном цикле деформирования.
Следовательно, площадь петли пропорциональна разнице между работой, затраченной на деформирование при нагружении и возвращенной при сокращении образца. Чем больше площадь петли, тем больше механической работы теряется в цикле нагружение - разгружение, превращаясь в тепловую энергию, Как видно, площадь петли гистерезиса характеризует механические потери в полимере при его деформировании.
Если аморфный полимер находится в стеклообразном состоянии, то протекание
http://www.mitht.org
релаксационных процессов сильно затрудняется вследствие того, что малоинтенсивное тепловое движение структурных элементов не может вернуть макромолекулы в исходное (до деформирования) состояние. Если полимер полностью или частично закристаллизован или кристаллизуется в процессе деформации, то протекание релаксационных процессов также затрудняется вследствие резкого повышения межмолекулярного взаимодействия в кристаллических областях полимера. Кристаллические структуры долго сохраняют свою устойчивость, не разрушаясь вследствие низкой интенсивности теплового движения элементов структуры. Поэтому в стеклообразных и кристаллических полимерах релаксационные явления труднее наблюдать и описывать. Если же в таких полимерах преобладают упругие гуковские деформации, то протеканием релаксационных процессов можно пренебречь при рассмотрении механических свойств этих полимеров. Поскольку при гуковских деформациях почти не изменяется положение элементов структуры; если же изменение все-таки происходит, то этот процесс их перемещения протекает очень быстро. Поэтому релаксационные процессы в таких полимерах тоже протекают быстро и их влияние на основные закономерности деформации очень мало.
В вязкотекучем состоянии релаксационные процессы также очень существенны и приводят к изменению формы, например, струи экструдата с течением времени. Неправильный учет их может привести к существенным нарушениям технологического процесса переработки термопластичных полимеров.
9. Механические свойства полимеров в стеклообразном состоянии.
По мере понижения температуры полимера, находящегося в высокоэластическом состоянии, интенсивность поступательных, а затем и колебательных движений его сегментов понижается, возрастает вязкость системы (межмолекулярное взаимодействие) и уменьшается ее удельный свободный объем. При достижении последним значения около 2,5 % движение сегментов практически прекращается и полимер становится стеклообразным - это происходит при температуре стеклования Тс. Этот процесс называется структурным стеклованием. В то же время, способность эластомера к высокоэластической деформации понижается при уменьшении времени действия механической нагрузки: если время ее действия меньше или равно времени релаксации, то полимер выше температуры структурного стеклования становится как бы твердым, т. е. в этом случае наблюдается «механическое стеклование» системы - подвижность сегментов полностью не исчезает, их расположение в пространстве не фиксируется, как при структурном стекловании, однако, скорость теплового движения оказывается меньше скорости приложения силы, и полимер ведет себя как стеклообразный. Следовательно, механическое стеклование наступает в высокоэластическом полимере. В дальнейшее речь пойдет о структурно-застеклованных полимерах.
Стеклование полимера связано с увеличением времени релаксации. При этом фазовый переход, т. е. качественная перестройка структуры, отсутствует. Совершенно очевидно, что химическая природа полимера - его полярность или неполярность должна существенно влиять на процесс стеклования, так как от нее зависят энергия межмолекулярного взаимодействия и возможность перемещения сегментов. У полярных полимеров более высокая энергия межмолекулярных взаимодействий элементов структуры, поэтому при снижении температуры подвижность сегментов уменьшается быстрее, а, следовательно, и стеклование наступает раньше, чем у неполярных. Действительно, с увеличением полярности температура стеклования полимеров возрастает; например, температура стеклования полиизобутилена или натурального каучука (гибкие молекулы, малое межмолекулярное взаимодействие) около -70 0С, у поливинилхлорида (высокое межмолекулярное взаимодействие) уже +80 0С, а у целлюлозы (жесткие макромолекулы, высокое межмолекулярное взаимодействие) температура стеклования лежит выше температуры химического разложения, т. е. этот полимер находится только в стеклообразном состоянии.
http://www.mitht.org
Возрастание времени релаксации перегруппировок сегментов и резкое понижение интенсивности их движений в области Тс приводит к тому, что при стекловании фиксируется неравновесная конформационная структура макромолекул: как следствие свойства стеклообразного полимера могут изменяться во времени. Чем быстрее происходит охлаждение при стекловании, тем в большей степени неравновесной оказывается структура стеклообразного полимера. Выдержка такого полимера при температуре, близкой к Тс, способствует постепенному протеканию необходимых релаксационных процессов и формированию более близких к равновесным полимерных стекол.
В связи с длинноцепочечным строением полимерных молекул и большим размером теряющих при стекловании подвижность кинетических элементов (сегменты) полимерные стекла, как правило, являются менее плотно упакованными, нежели их низкомолекулярные аналоги, и имеют больший свободный объем.
Доля свободного объема в стеклообразных полимерах связана с химическим строением макромолекул, т. е. с их гибкостью. Чем меньше размер сегмента, тем легче осуществляются при стекловании необходимые перемещения сегментов, тем более плотно упакованной и близкой к равновесной оказывается конформационная структура цепей. И наоборот, полимеры с жесткими макромолекулами образуют более рыхло упакованныe системы; в связи с огромными временами релаксации неравновесность таких систем сохраняется сколь угодно долго - эти системы принято называть метастабильными.
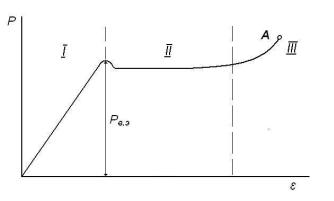
При воздействии постепенно возрастающих механических нагрузок стеклообразные полимеры сначала деформируются как обычные твердые тела (участок I на рис. 13): деформация носит обычный упругий характер, т. е. обусловлена изменениями валентных углов, длин связей и межмолекулярных расстояний. На участке II при практически постоянной нагрузке наблюдается значительное (до нескольких десятков процентов) удлинение полимера, визуально проявляющееся в образовании на образце более тонкой части («шейки») и ее постепенном удлинении. Участок III отвечает деформации материала шейки до её разрыва (точка А). Если растяжение прекратить незадолго до разрыва, то деформации, развившиеся на участках I и III, исчезнут, а на участке II сохранятся. Однако после нагревания материала растянутой шейки до Тс или чуть выше эта деформация также исчезнет - образец примет первоначальные размеры. Указанный факт свидетельствует о высокоэластическом характере (большая по величине и обратимая по природе) деформации, сопровождающей образование шейки.
Рис. 13. Зависимость напряжение – деформация для аморфного стеклообразного полимера; Pв.э. – предел вынужденной эластичности.
Высокоэластические деформации, развивающиеся в стеклообразных полимерах под действием достаточно больших механических нагрузок (10-200 МПа в зависимости от температуры и природы полимера), называют вынуждено-эластическими. Механизм их развития тот же, что и обычных высокоэластических – конформационные превращения макромолекул за счет перемещений сегментов. Однако ниже Тс они перемещаются не самопроизвольно вследствие теплового движения, а вынужденно - под действием механической нагрузки. После снятия нагрузки исчезнет только упругая деформация (доли %), т.к. тепловое движение не может вернуть растянутые макромолекулы в исходные конформационные состояния и растянутый на стадии формирования шейки образец
http://www.mitht.org
сохраняет удлиненную форму: в нем как бы «заморожены» новые более вытянутые конформации цепей. Однако, при нагревании выше Тс сегменты обретают вновь способность к тепловым перемещениям и образец сокращается до длины близкой к исходной.
Если стеклообразный полимер деформирован на определенную величину, меньшую, чем деформация, соответствующая пределу вынужденной эластичности, то релаксация напряжения незначительна, и напряжение, возникшее при заданной деформации, сохраняется. Образец (изделие) сохраняет размеры и формы под нагрузкой. Это отличает стеклообразные полимеры от эластомеров и позволяет применять их в качестве конструкционных материалов, детали из которых, работают в условиях заданных деформаций или напряжений.
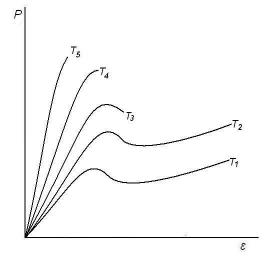
Напряжение, при котором в стеклообразном полимере развивается вынужденная эластическая деформация, называют напряжением вынужденной эластичности. При понижении температуры величина предела вынужденной эластичности увеличивается, а область её сокращается вплоть до полного исчезновения, когда разрушение образца наступает до достижения предела вынужденной эластичности (рис. 14). При понижении температуры тепловая энергия сегментов снижается и не может преодолеть силы межмолекулярного взаимодействия, что необходимо для развития высокоэластической деформации. Поэтому предел вынужденной эластичности возрастает. Температура, при которой разрушение образца совпадает с пределом вынужденной эластичности, называется
температурой хрупкости Тхр (Т4 на рис. 14). Ниже этой температуры (Т5)
вынужденноэластические деформации не развиваются, и полимер находится только в хрупком состоянии. Хрупкость – это способность стеклообразных полимеров разрушаться при малых деформациях, меньших, чем деформация, соответствующая пределу вынужденной эластичности.
Рис. 14. Влияние температуры на вид кривых напряжение – деформация для аморфного стеклообразного полимера (Т1 >Т2 >Т3 >Т4 >Т5).
Интервал Тс - Тхр является той областью температур, в которой разрушение стеклообразного полимера сопровождается значительной деформацией, т. е. является нехрупким. Обычно температура хрупкости определяет нижний температурный предел использования полимеров как твердых пластиков.
Полимеры с гибкими цепями образуют, как правило, плотно упакованные стекла, обладающие незначительными возможностями для проявления вынужденной эластичности и, следовательно, небольшим интервалом нехрупкого состояния. Наоборот, для рыхлоупакованных полимерных стекол на основе макромолекул средней жесткости сохраняются возможности вынужденного перемещения сегментов под действием механической нагрузки до достаточно низких температур.
Кроме химического строения цепей, определяющего их гибкость, величина нехрупкого интервала в случае олигомеров может определяться и молекулярной массой, для
http://www.mitht.org
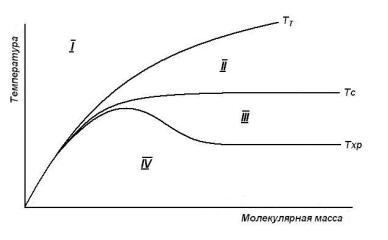
достаточно высокомолекулярных образцов температура хрупкости (как и Тс) от длины цепей не зависит.
Рис. 15. Зависимость от молекулярной массы температур текучести, стеклования и хрупкости: I – вязкотекучее состояние; II – высокоэластическое состояние; III – вынужденноэластическое состояние; IV – хрупкое состояние.
Кривые рис. 15 являются наглядной характеристикой взаимосвязи между молекулярной массой аморфного полимера и температурными областями их релаксационных состояний, при этом кривая Тхр разделяет область стеклообразного состояния полимера на области нехрупкого (вынужденно-эластического) и хрупкого состояний.
Температура хрупкости определяет морозостойкость полимеров в стеклообразном состоянии. Методы определения морозостойкости - это, как правило, методы определения той температуры, при которой полимер начинает хрупко разрушаться. Так, полимер в виде бруска, закрепленного консольно, охлаждают, определяя температуру, при которой он разрушается под действием заданного груза, падающего на него. Другой способ, применяющийся для пленочных материалов, состоит в том, что пленку сгибают в виде петли и охлаждают. Температура, при которой сплющивание петли приводит к излому пленки, характеризует морозостойкость пленки. Все методы определения морозостойкости так или иначе состоят в определении температуры, при которой полимер хрупко разрушается либо в условиях действия нагрузки заданной величины, либо деформирования на заданную величину.
Полимер становится хрупким тогда, когда время до разрушения много меньше, чем время релаксации, и поэтому никакой перегруппировки сегментов под действием силы не происходит. Это и определяет незначительную величину деформации при разрушении. Вынужденно-эластические деформации в хрупких полимерах развиться не успевают, но вследствие наличия остаточного свободного объема в стеклообразном полимере его хрупкое разрушение происходит при деформации около 1 % (или немного больше), в то время как силикатные стекла разрушаются при деформации 0,1 %. Ценность стеклообразных полимеров в первую очередь определяется именно тем, что они обладают пониженной хрупкостью по сравнению с силикатным стеклом, т. е. большим сопротивлением разрушению при ударе.
Снижению хрупкости полимерных стекол способствует то, что при Т < Тс в них самопроизвольно происходят так называемые вторичные релаксационные переходы, связанные с перемещением молекулярных группировок, меньших, чем размер сегмента. Это приводит к диссипации энергии, в том числе и энергии удара, и делает полимерные стекла существенно более стойкими к удару по сравнению с низкомолекулярными силикатными стеклами.
10. Особенности механических свойств в кристаллических полимерах.
http://www.mitht.org