

конъюгаты с фс
.pdfBioconjugate Chemistry |
|
REVIEW |
conjugates.136 A distribution of attachment points results in a distribution of orientation in NP-protein conjugates, and thus a distribution of protein activity or mixed avidity. Furthermore, the reactivity of many di erent amino acid residues can potentially result in undesirable side-reactions. It is generally the case that proteins have a comparable number of carboxyl (e.g., glutamic acid, aspartic acid) and lysine residues. Thus, protein protein cross-linking e ciently competes with NP-protein coupling when using EDC chemistry. Multiple lysine residues can also drive proteinmediated NP-NP cross-linking. Such side reactions only increase the challenge of controlling bioconjugate valence and orientation, while introducing additional heterogeneity across an ensemble.
In contrast to proteins, it is often possible to design synthetic oligonucleotides and oligopeptides that are monoreactive. While this allows for control over the attachment, it does not necessarily guarantee orientation. Flexible linkages allow the biomolecules in NP-conjugates to adopt conformations that interact with the surface of the NP. For example, it has been shown that oligonucleotides can strongly adsorb to NPs such as CdSe/ZnS QDs,137 gold NPs,138 and carbon nanotubes.139 It is critical to note that the interactions between a NP and a biomolecule in a conjugate are a function of the combination of the NP, its interfacial chemistry, and the biomolecule. Therefore, e orts to control the display of biomolecules on NPs must consider these factors in addition to the details of the coupling chemistry.
There are many applications in biosensing that require good control over conjugate valence and biomolecule orientation. As noted earlier, additional complexity is associated with these applications due to the frequent tendency toward “nested” conjugate preparation. That is, an optically active NP is conjugated with a biomolecule, and that biomolecule is also conjugated with a small molecule that acts as a reporter. Good examples are QD probes for protease activity. A peptide sequence cleaved by a target protease is connected to a QD at one terminus and a fluorescent dye at its other terminus. The peptide creates the proximity required for F€orster resonance energy transfer (FRET), and this proximity is lost as a consequence
of target protease activity, thereby generating an optical signal for transduction.41,42,140 The important point is that FRET
is sensitive to both the acceptor ratio and separation distance between the QD and dye in this configuration. Therefore, two-fold control over the preparation of the NP-bioconjugate is required for optimal function in this type of sensing.
There are also challenges associated with the preparation of NP-small molecule conjugates. Depending on the application, control over conjugate valence may not be critical. In the case of drug delivery, the intent is often to “load up” the NP to concentrate delivery. Providing that NP solubility is not compromised, shotgun-labeling strategies can thus be e ective for preparing NP-drug conjugates. Many drugs and other small molecules can also be synthesized with a uniquely reactive group, used in excess in conjugate reactions, and are amenable to the e cient purification of NP-conjugates. These factors o er a degree of control that is often more di cult to achieve with large biomolecules. However, the relatively small size of drugs and other small molecules compared to NPs can also be a potential challenge. The mode of action of many drugs requires interaction with, for example, the binding site of a protein. NPconjugated drug molecules can have availability for this interaction, but it must be mediated through long and flexible linkers, or triggered release of the drug from the NP under suitable conditions.
Overall, the controlled display of biomolecules on NPs is not a trivial feat. Although the optimization of most experiments will not necessarily require satisfying all of the ideal criteria discussed above, most would benefit tremendously by the development and use of bioconjugate chemistries that come close to satisfying a majority of these criteria.
Bioorthogonality. The example of EDC bioconjugate chemistry is a notorious one: it is both widely used and has several liabilities. However, there are many other standard bioconjugation chemistries that have, at least to some degree, similar liabilities. For example, isothiocyanate derivatives of biomolecules are amine reactive and stable in aqueous solution, potentially providing a greater degree of control. However, the resulting thiourea linkage can degrade,141 and a major limitation is still the ubiquitous nature of lysine residues in proteins. In contrast, cysteine residues are scarce in proteins, and bioconjugate chemistries that target the reactive thiol of cysteine residues can potentially ameliorate many of the liabilities associated with EDC. For example, maleimides hydrolyze more slowly than o-acylisourea intermediates, and while the latter are reactive toward all good nucleophiles (e.g., amine, thiol, hydrazide), the reaction of maleimides with thiols is highly selective at nearneutral pH. This selectivity, combined with the ability to recombinantly engineer unique cysteine residues into proteins and synthetically insert them into peptides, can provide a welldefined point of attachment between a biomolecule and a maleimide-activated NP. This enables a degree of control over biomolecular orientation and largely avoids issues associated with undesirable cross-linking or other side reactions.
The idea of highly selective or specific bioconjugate chemistry reactions is captured in the concept of “bioorthogonality.”142 144 Bioconjugate chemistries that are bioorthogonal are those that do not have significant reactivity toward the functional groups that are intrinsic to biomolecules. This generally encompasses amine, carboxyl, hydroxyl, and thiol groups. However, to be strictly bioorthogonal, the chemistry must also be unreactive toward alkenes, amides, disulfides, esters, phosphodiesters, and a plethora of other functional groups that can be found within the biological milieu. The best candidates for strictly bioorthogonal reactions appear to be cycloaddition reactions from click chemistry. These reactions can enable clean and e cient labeling in biological matrices as complex as serum.145 Other reactions, such as that between a hydrazide and an aldehyde/ketone (hydrazone ligation), are not strictly bioorthogonal, but can be e ectively bioorthogonal if biological aldehydes/ketones are not native to a particular experimental matrix of interest. Several reactions with bioorthogonal character are discussed in detail later. An important caveat to bioorthogonal chemistries is that the functional groups providing the high reaction specificity are generally not naturally occurring. In the case of synthetic oligonucleotides and oligopeptides, incorporating a moiety with the necessary functional group(s) is readily accomplished during synthesis. However, in the case of proteins, the use of bioorthogonal chemistry may require a nested bioconjugate reaction using more traditional labeling chemistry. A common strategy is the use of heterobifunctional cross-linkers with a non-bioorthogonal reactive moiety (e.g., succinimidyl ester, maleimide) that introduces the desired bioorthogonal functional group(s) of interest through shotgun or site-directed labeling methods. An alternative to heterobifunctional cross-linkers is the incorporation of unnatural amino acids into proteins. These unnatural amino acids can have, for example, an azide or alkyne functionality on the side chain.146
835 |
dx.doi.org/10.1021/bc200065z |Bioconjugate Chem. 2011, 22, 825–858 |
Bioconjugate Chemistry |
|
REVIEW |
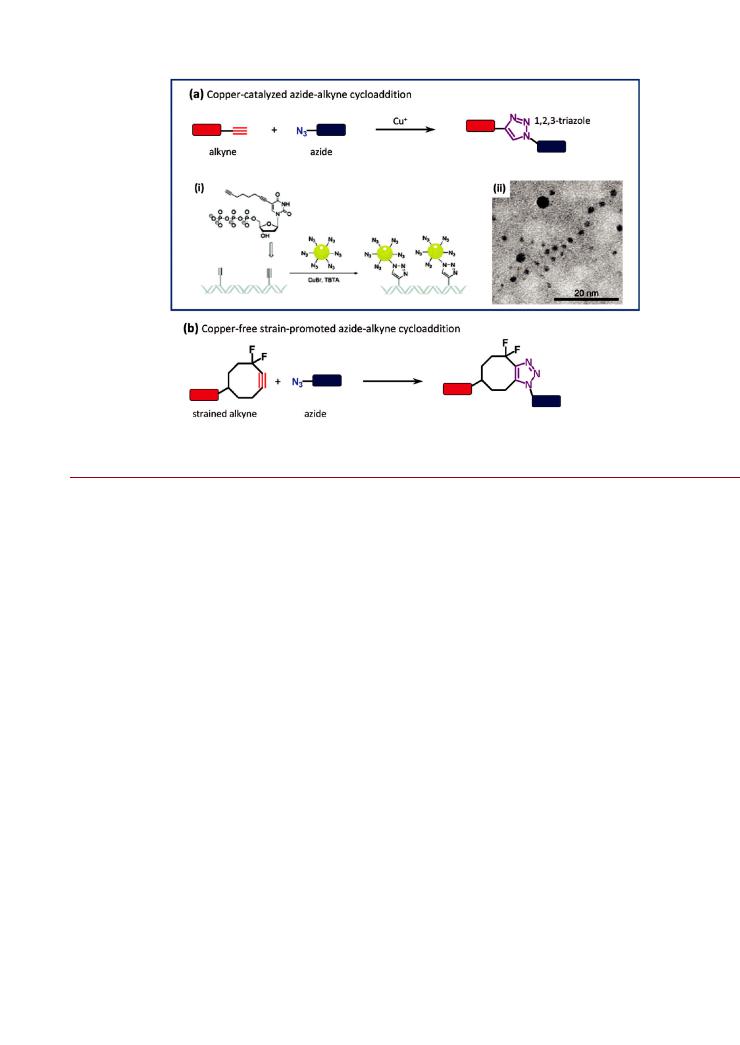
Figure 8. Azide alkyne cycloaddition (AAC) reactions: (a) copper-catalyzed (CuAAC); (b) copper-free/strain-promoted. The example of the CuAAC shows (i) the use of alkyne-modified thymidine bases along double-stranded DNA to align azide-modified Au NPs. An electron microscope image of the aligned Au NPs is shown in (ii). Part of (a) is reproduced from ref 168. Copyright 2008 Royal Society of Chemistry.
Most NP-bioconjugates are prepared in clean matrices (e.g., bu ers) and subsequently applied in a biological matrix. Bioorthogonal chemistry is therefore important because it potentially o ers some control over NP bioconjugation by providing a well-defined point of attachment between the NP and the biomolecule, and does so without undesirable side reactions. Furthermore, bioorthogonal chemistries can facilitate nested conjugate reactions with control at each step. For example, a peptide can be coupled to a NP through a bioorthogonal reaction and concurrently labeled with a dye or other reporter through more traditional methods at lysine or cysteine residues. Similarly, the use of bioorthogonal chemistries also potentiates the controlled attachment of multiple biomolecules to a NP—for example, two di erent peptide sequences, a mixture of a peptide and an oligonucleotide, an antibody and a drug molecule, or other combinations of biomolecules that impart multifunctionality to a NP. This could take the form of two distinct bioorthogonal chemistries that are applied concurrently or in succession to conjugate a NP with two di erent biomolecules, and where the biomolecule orientations and conjugate valences are controlled and biological activities are not compromised. Such a level of control is extremely challenging to achieve using bioconjugate chemistries wherein two biomolecules compete for common reaction sites at a NP or have potential side-reactions with one another. The development of multifunctional NP-bioconjugates thus stands to benefit immensely from bioorthogonal reactions— particularly combinations that are also mutually orthogonal.
Bioorthogonality is just one important part of the toolkit that researchers are developing for the preparation of NP-bioconju- gates. Another important part of the toolkit is reaction e ciency. Ideal conjugate chemistries use reactants that are stable in aqueous solution, proceed e ciently at 1:1 stoichiometries and at low concentrations, and have short reaction times at ambient
or physiological temperature in aqueous media. Many of the chemistries that satisfy these criteria do not use an active crosslinker (e.g., EDC), but rather make use of a catalyst or strained bonds to drive the coupling reaction. Overall, the controlled display of biomolecules on NPs requires three ingredients: bioorthogonal reactivity, high reaction e ciency, and controlled NP properties.
’ADVANCES IN NANOPARTICLE BIOCONJUGATION CHEMISTRY
Covalent Chemistries. Cycloaddition Reactions. A cycloaddition is a reaction where two unsaturated molecules combine to form a cyclic adduct in which the number of σ bonds increases.147 Since biological macromolecues rarely contain moieties with nonaromatic double bonds, cycloaddition strategies have significant potential for bioorthogonality and have found increasing utility for the selective modification of NPs. The Huisgen 1,3-dipolar cycloaddition, illustrated in Figure 8a, is a reaction between an alkyne and an azide that produces a triazole linkage. In the presence of a copper(I) salt, the reaction is greatly accelerated, regioselective, and highly efficient. Referred to as the “copper-catalyzed azide alkyne cycloaddition” (CuAAC), and well-known as the flagship reaction of the “click” chemistry family, this reaction has become a valuable ligation method for the preparation of bioconjugates. The CuAAC reaction is advantageous in that it is bioorthogonal and proceeds under relatively mild conditions in aqueous solution. In addition to being used in
drug synthesis, controlled surface modification, protein labeling, and activity-based protein profiling,148,149 the CuAAC has also
been used to control the display of biomolecules on NPs. Viral capsid bioconjugates were one of the first NP systems to
which CuAAC chemistry was extensively applied. Bioconjugates
836 |
dx.doi.org/10.1021/bc200065z |Bioconjugate Chem. 2011, 22, 825–858 |

Bioconjugate Chemistry |
|
REVIEW |
of this naturally occurring biological NP family have been prepared
using CuAAC with fluorescent dyes,150 155 MRI contrast agents,154,156,157 biotin,152,154 linear and branched polysaccharides,150,155,158 160 peptides,155,161 and proteins.141,155 In most
cases, the initial alkyne or azide functional group is introduced onto the virus capsid using a succinimidyl ester derivative that couples to available lysine residues. For example, CPMV was modified with an NHS-alkyne derivative and subsequently “clicked” with an azide-terminated PEG-folate conjugate for tumor cell targeting experiments.162 Both CPMV and bacteriophage Qβ have been modified with lanthanide complexes to
explore the biodistribution, toxicology, and pathology of viral particles in mice.157,163 Here, the CPMV was modified with an
NHS-azide derivative and clicked with an alkyne derivative of a Gd3þ-tetraazacyclododecanetetraacetic acid chelate. NP hybrids composed of C60 labeled Qβ bacteriophage particles have also been prepared using CuAAC chemistry.164 It was estimated that 30 40 C60 molecules were attached per Qβ particle, with the final NP hybrids showing uptake in HeLa cells.
The CuAAC reaction has also been used to prepare controlled bioconjugates with a variety of inorganic NP materials. Examples
include the enzymes trypsin, luciferase, and lipase being conjugated to Au NPs.165 167 In the former, Au NPs were coated
with a carboxyl-functionalized polymer, reacted with an amine- PEG-azide linker using EDC activation, and clicked with alkyne labeled trypsin. The trypsin was labeled via the reaction of its lysine residues with EDC activated 4-pentynoic acid. The activity of trypsin-Au NP conjugates prepared in this manner was 3-fold higher than analogous conjugates prepared using electrostatic adsorption or direct EDC coupling between the lysine residues and Au NPs.165 As shown in Figure 8a (i) and (ii), alkyne modified nucleobases have also been used for the controlled alignment of azide functionalized Au NPs along a DNA strand at regular intervals.168 Magnetic Fe3O4 NPs coated with silica/aminosilane have been functionalized with alkyne or azide groups and then clicked to display monosaccharides and disaccharides, the Tn antigen, the flag peptide, biotin, human serum albumin, and maltose binding protein (MBP).169 171 Silica NPs have also been clicked to form conjugates with fluorescent dyes and bovine serum albumin.172 In addition to the use of cross-linker chemistry to introduce azide or alkyne groups to the NPs with conventional functional groups
(e.g., carboxyl, amine), azide and alkyne derivatives of silanes have been synthesized and used to coat NPs.171,173 The CuAAC
has been further applied to the preparation of fullerene174 and carbon nanotube175 177 bioconjugates with carbohydrates, porphyrins, and phthalocyanine derivatives. The alkyne functionality was introduced to CNTs through a diazonium salt, and introduced to C60 through a cyclopropanation reaction. Numerous reports detail the use of the CuAAC reaction for the synthesis of dendrimers, polymers, polymeric micelles, and polymersomes. These studies and the preparation of bioconjugates can be found in other reviews.178 180
The major limitation of the CuAAC reaction is the requirement of a Cuþ catalyst. In cellular and protein-based applications, the cytotoxicity of Cuþ and its ability to mediate protein modifications are potentially limiting. Although much of the undesired reactivity can be mitigated through the use of copper ligands and organic scavengers,181 the use of a Cuþ catalyst is nonetheless poorly suited to some experimental systems. To address this challenge, the Bertozzi group pioneered the use of strain-promoted AAC as an alternative ligation route that does not require a catalyst—“copper-free click chemistry”. As illustrated in
Figure 8b, the copper catalyst is avoided by using an alkyne in a
strained geometry, such as by inclusion in a cyclooctyne-based ring system.182,183 The copper-free AAC approach has already
been utilized in several biological studies and applications.184 187 However, it should be noted that the strained alkyne moiety is intrinsically more reactive toward other nucleophiles, and thus, chemoselectivity is potentially reduced in some systems. The application of the copper-free AAC reaction in the preparation of NP-bioconjugates has been limited, likely due to challenges associated with solubility and the multistep synthesis of the strained cyclooctyne structures. One of the few examples is the conjugation of QDs with mannosamine.188 The use of the strainpromoted AAC was motivated by the irreversible PL quenching e ect copper ions have on CdSe/ZnS QDs, and utilized an azidoderivative of mannosamine in combination with amine-modified QDs that were reacted with a carboxyl-derivative of cyclooctyne via EDC. In another example, chitosan-g-PEG NPs with available azide groups were conjugated to antibodies that had been labeled with a NHS-PEG-cyclooctyne derivative.189 In this case, depolymerization initiated by the presence of Cuþ motivated the use of the strain-promoted AAC.
In addition to reactions between an azide and an alkyne, cycloaddition reactions can occur between tetrazine and strained double bonds, such as those in trans-cyclooctene or norbornene, via a [4 þ 2] cycloaddition. Tetrazine ligations are inverse- electron-demand Diels Alder reactions that proceed rapidly (up to 2000 M 1 s 1) without a catalyst, and with nearly 100% conversion at micromolar reactant concentrations and room temperature.190 The reaction, illustrated in Figure 9a, is bioorthogonal and produces only nitrogen as a byproduct. Due to its relative novelty, application of tetrazine ligation to NPs has been more limited than CuAAC. Han et al. prepared QDepidermal growth factor (EGF) conjugates using the tetrazine ligation,191 where EGF was labeled with a succinimidyl ester derivative of 3-phenyl-1,2,4,5-tetrazine, and a QD coated with an amine-functionalized polymer was modified with a succinimidyl ester derivative of norbornene. The QD-EGF conjugates were prepared in vitro and subsequently used to label cells that expressed EGF receptor in vivo. The bioorthogonality of the chemistry enabled in situ conjugation, where cells were first incubated with tetrazine labeled EGF and subsequently exposed to norbornene-modified QDs for labeling. The tetrazine ligation has been used similarly by Haun et al. to prepare dye-labeled magnetic NP-antibody conjugates.192 In this case, the strained alkene was a succinimidyl ester derivative of trans-cyclooctene that was used to label an anti-EGF receptor antibody rather than the NP. Amino-functionalized and dye-labeled magnetic NPs were modified with a succinimidyl ester derivative of 3-phenyl- 1,2,4,5-tetrazine. The ligation reaction was then applied to cellular labeling, and could be done in vitro prior to cellular labeling, or in situ after labeling cells with antibody-cyclooctene conjugates.
Similar to the tetrazine ligation, Shi et al. have used a [4 þ 2] cycloaddition reaction between a maleimide-labeled antibody and furan groups associated with the corona of poly(2-methyl- 2-carboxytrimethylene-carbonate-co-D,L-lactide)-graft-poly- (ethylene glycol)-furan biodegradable micellular NPs.193 The chemistry is illustrated in Figure 9b. The maleimide was introduced site-specifically at the carbohydrate chains within the Fc region of the anti-HER2 antibodies (targeting breast cancer cells). This cycloaddition reaction was utilzed due to its selectivity and e ciency under mild conditions to help preserve antibody activity and avoid cross-linking reactions. The bioorthogonality
837 |
dx.doi.org/10.1021/bc200065z |Bioconjugate Chem. 2011, 22, 825–858 |

Bioconjugate Chemistry |
|
REVIEW |
Figure 9. Cycloaddition reactions: (a) tetrazine ligation; (b) Diels Alder reaction. The example of the tetrazine ligation shows (i) the labeling of epidermal growth factor (EGF) with a succinimidyl ester derivative of 1,2,4,5-tetrazine, and (ii) two-step labeling of cells with norbornene-modified QDs. Fluorescence and di erential interference contrast (DIC) images of labeled cells are shown in (iii). Part of (a) is adapted with permission from ref 191. Copyright 2010 American Chemical Society.
of this chemistry requires the absence of other nontargeted thiol groups and control of pH to prevent the undesired reaction of amines with the maleimide.
Staudinger Ligation. Originally described in 1919, the Staudinger reduction is the reaction of a terminal azide with a phosphine to produce an aza-ylide intermediate that hydrolyzes under aqueous conditions to produce a primary amine.194 In 2000, Saxon and Bertozzi developed a modified “Staudinger ligation,” shown in Figure 10a, where the aza-ylide is trapped by an adjacent electrophilic carbonyl group to yield an amide bond after hydrolysis.195 As a consequence of its bioorthogonality and chemoselectivity, the Staudinger ligation has been extensively used for joining or labeling biomolecules with applications that include the fluorescent labeling of proteins and DNA, protein immobilization to surfaces, and specific labeling of unnatural glycoproteins on cellsurfaces.196 The shortcoming of this chemistry is the potential oxidation of phosphines under ambient conditions or by metabolic enzymes, which is commonly overcome through the use of excess reagents. It should also be noted that “traceless” Staudinger ligations
have been developed, where the phosphine oxide is cleaved from the ligated intermediate during hydrolysis to form an amide.197,198
Although the Staudinger ligation has been used with biomolecules in several studies, there have been only a few reports of the preparation of NP-bioconjugates. Zhang et al. used the Staudinger ligation to prepare liposome lactose conjugates,199 where the liposomes incorporated a phospholipid with a triphenylphosphine modification of its headgroup. The Staudinger ligation has also been used to attach RGD peptides to polyamidoamine-DNA NPs for improved cellular uptake of the NPs.200
Hydrazone and Oxime Ligations. The reaction between hydrazino groups (or hydrazido groups) and carbonyls (e.g., aldehydes or ketones) yields hydrazones as illustrated in Figure 10b. Although the reaction is reversible, the equilibrium favors the hydrazone under aqueous conditions (Keq = 104 106 M 1).201 Forward and reverse reaction rates are slow at neutral
pH but can be accelerated at acidic pH or with the use aniline as a catalyst.201,202 The latter enables fast and efficient ligation
(minutes to hours) at neutral pH and, since it is typically straightforward to remove aniline from the bioconjugates following the reaction, can yield relatively stable linkages due to the slow uncatalyzed rate of hydrolysis at neutral pH (10 6 s 1).201 In addition, hydrazone ligation is chemoselective and potentially bioorthogonal to the functional groups in proteins, oligonucleotides, and carbohydrates—the exception being reducing ends of the latter. As such, it is another attractive method for the preparing NP-bioconjugates.
Brunel et al. modified the lysine residues of CPMV NPs using sulfo-succinimidyl 4-formylbenzoate, and subsequently decorated the NP with two di erent hydrazido-terminated synthetic peptide sequences using hydrazone ligation.203 The modified CPMV NP was then used as an imaging probe for cells and tumor xenografts, where one peptide was used for targeting and the other was modified with PEG for biocompatibility and fluorescently labeled for tracking. In a similar strategy, the use of
arylhydrazino-terminated peptides has also been applied to the preparation of QD-bioconjugates.204,205 As shown in Figure 10b,
a “starter” peptide displaying a C-terminal hexahistidine sequence that enabled self-assembly onto CdSe/ZnS QDs (vide infra) and
838 |
dx.doi.org/10.1021/bc200065z |Bioconjugate Chem. 2011, 22, 825–858 |
Bioconjugate Chemistry |
|
REVIEW |
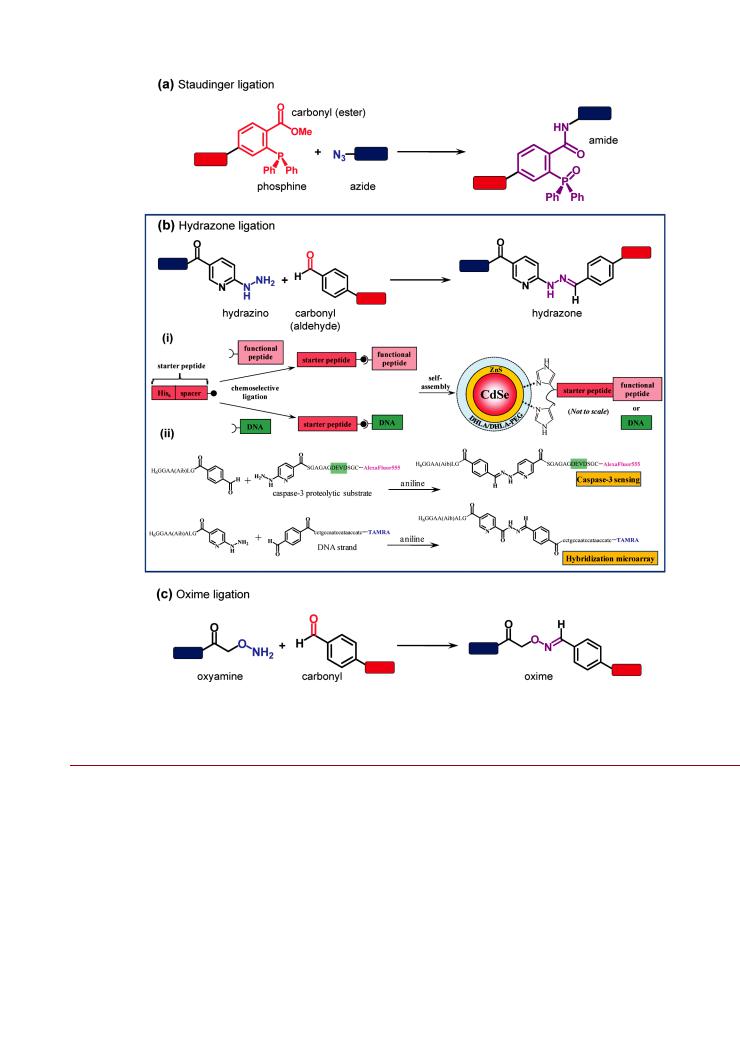
Figure 10. Chemoselective ligations: (a) the Staudinger ligation; (b) hydrazone ligation; (c) oxime ligation. The example for the hydrazone ligation (b) shows (i) a strategy for the modular assembly of functional peptides or DNA on QDs using a terminal polyhistidine “starter peptide” modified with a HYNIC or benzaldehyde group at the opposite terminus. The functional peptide or DNA is modified with the complementary functionality for hydrazone ligation (benzaldehyde or HYNIC, respectively). Two examples of modular assembly are shown in (ii), and include a peptide substrate for proteolytic monitoring and a DNA probe. Part of (b) is adapted with permission from ref 204. Copyright 2010 American Chemical Society.
an N-terminal 4-formylbenzoyl group was the basis for this strategy. Aniline-catalyzed hydrazone ligation between the starter peptide and another peptide or oligonucleotide modified with a terminal 2-hydrazinonicotinoyl group (HYNIC) yielded cell-penetrating peptides, protease substrates, or hybridization probes that were subsequently self-assembled to the QD to yield the final active bioconjugate. The nucleophilic aniline catalyst increased the conjugation rate and enabled the coupling reaction to proceed without an excess of hydrazine peptide (cf. Brunel
et al.203). The hydrazone ligation could be done prior to or after in situ assembly of the starter peptide on the QD.204,205
Rather than being considered a potential liability, the pHsensitivity of the hydrazone bond may actually be attractive for use in drug delivery. In the physiological pH range between 7.3
and 7.5, the hydrazone bond hydrolyzes very slowly and is thus stable over the time course of many experiments. However, the hydrolysis of the hydrazone bond becomes appreciable at the slightly acidic pH range (pH 5 6) associated with endosomes
and lysosomes. As a consequence, doxorubicin has been conjugated to magnetic NPs206,207 and Au NPs208,209 through
hydrazone linkages for targeted NP-drug conjugate delivery at physiological pH and subsequent drug release following endocytic cellular uptake. An analogous conjugation strategy has also been used with a cis-platin prodrug and polymer NPs.210 In these studies, the hydrazido group was introduced to the NP through chemical modification of its stabilizing ligands or polymer coating. This included the hydrazinolysis of ester groups,207 209 and the reaction of carboxyl groups with adipic acid dihydrazide via
839 |
dx.doi.org/10.1021/bc200065z |Bioconjugate Chem. 2011, 22, 825–858 |
Bioconjugate Chemistry |
|
REVIEW |
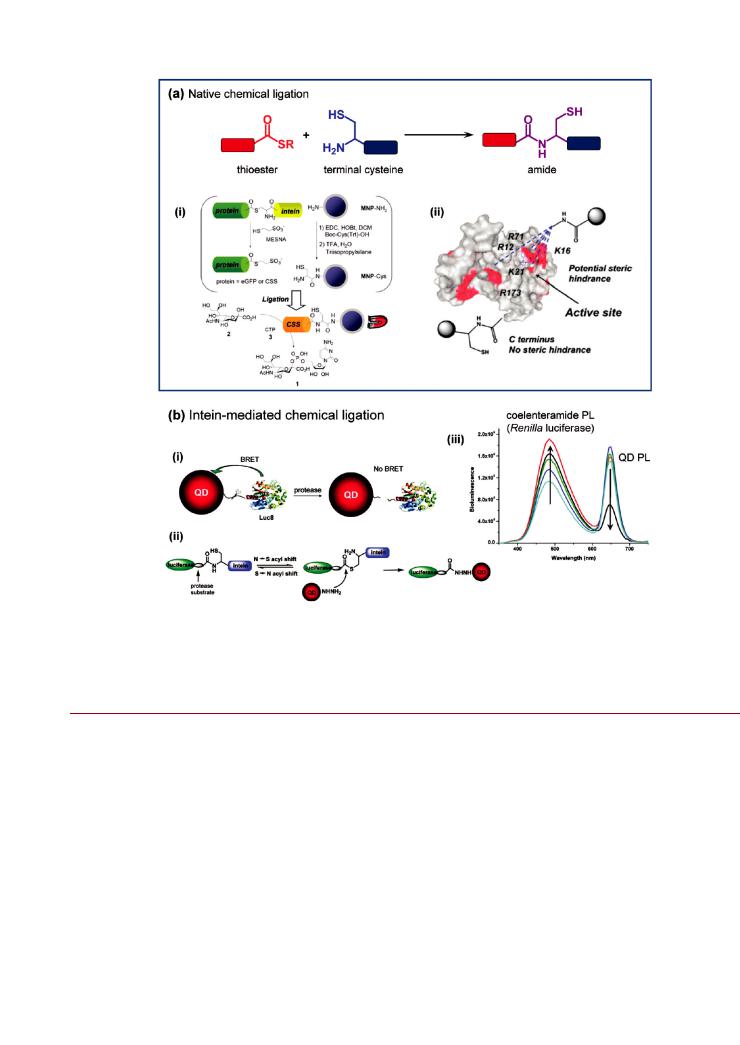
Figure 11. (a) Native chemical ligation between a thioester and a terminal cysteine. The example shows (i) enzyme (CSS) bioconjugate preparation using an intein to generate a thioester for native chemical ligation to a cysteine modified magnetic NP; and (ii) a comparison of the steric hindrance at the active site of CSS between labeling at the C-terminus via native chemical ligation and labeling at lysine (K) or arginine (R) residues. (b) Intein-mediated chemical ligation. The example shows (i) a proteolytic sensing strategy using QD-luciferase conjugates and BRET, (ii) the corresponding inteinmediated ligation chemistry, and (iii) loss of BRET between the excited state Renilla luciferase product (coelenteramide) and QDs. Increasing protease activity is in the direction of the arrows. Part of (a) is reproduced from ref 223. Copyright 2008 Royal Society of Chemistry. Figure in (b) are adapted with permission from ref 231. Copyright 2008 American Chemical Society.
carbodiimide coupling.206 In particular, doxorubicin facilitates use of the hydrazone bond strategy since it has an intrinsic ketone group for site-specific ligation with hydrazido-modified NPs.
An oxime is the product of the ligation reaction between an oxyamine and an aldehyde or a ketone (Figure 10c). Although an oxime is more stable toward hydrolysis than a hydrazone (Keq g
108 M 1), it is similarly characterized by slow reaction rates at neutral pH and e cient ligation requires catalysis by aniline.201,211
Oxime ligation has been used to prepare fluorescent dye and cellpenetrating peptide conjugates of polysaccharide NPs displaying a reducing chain end.212 Both the dye and the peptide were aminooxy-modified for these purposes. Similarly, glycan conjugates of Au NPs coated with bifunctional aminooxy-alkyl-thiol
ligands have been prepared via oxime ligation with the reducing ends of glycans.213,214 This strategy was motivated by the
observation of poor glycan-protein binding specificity when conjugates were prepared through reductive amination of the
glycan.215 In another example, the exterior lysine residues of capsids of bacteriophage MS2 were modified with succinimidyl 4-formylbenzoate and bound to a fluorescent dye synthetically
modified with a PEG linker that terminated in an oxyamine group.216
Native Chemical Ligation. The reaction between a peptide with an N-terminal cysteine and a second peptide with a C-terminal thioester to yield an amide bond is illustrated in Figure 11a, and is referred to as native chemical ligation (NCL).217 219 The reaction proceeds via transthioesterification, which links the peptides through an intermediate thioester that undergoes a spontaneous intramolecular SfN acyl shift rearrangement to form the amide and regenerate the cysteine side chain thiol. The reaction is highly chemoselective and does not require protection of any other amino acid side chains (including nonterminal cysteine residues, which do not form a stable amide). Furthermore, the C-terminal thioester may be associated
840 |
dx.doi.org/10.1021/bc200065z |Bioconjugate Chem. 2011, 22, 825–858 |

Bioconjugate Chemistry |
|
REVIEW |
with any amino acid. These properties combine to make NCL an attractive chemistry for linking peptides to other peptides, proteins, or other substrates.
NCL has been used to chemoselectively modify lipid micelles and liposomes with single domain antibodies and a recombinant collagen binding protein, CNA35.220 222 The phospholipids were modified at the headgroup with a cysteine terminated PEG chain, and a C-terminal thioester was introduced to the proteins using self-cleavable intein domains that were associated with the protein expression system. Similar expression techniques have been used with enhanced green fluorescent protein (eGFP) and CMP-sialic acid synthetase (CSS) to generate a C-terminal thioester that could be ligated with cysteine modified magnetic NPs (Figure 11a).223 The magnetic NPs were synthesized with an amine coating, coupled with protected cysteine using carbodiimide/hydroxybenzotriazole activation, and deprotected for the NCL reaction. The conjugation of CSS to the magnetic NPs using NCL yielded a much smaller decrease in activity (33%) compared to nonchemoselective coupling via suberic acid bis-N-hydroxysuccinimide ester cross-linker (77%). This was attributed to the random enzyme orientation associated with reaction of the cross-linker with arbitrary CSS amine groups. Becker et al. have also used NCL to control the orientation of enzymes in NP-bioconjugates.224 In this case, a guanosine triphosphate hydrolase (small GTPase) was expressed with an N-term- inal peptide sequence that was cleaved at a cysteine residue by a protease. A bifunctional linker was synthesized with a terminal thioester and protected terminal thiol, where the former facilitated ligation to the GTPase and the latter was deprotected for subsequent assembly on Au NPs.
Expressed Protein Ligation. Biological expression of C-term- inal thioester or N-terminal cysteine peptides enable the NCL strategy to be applied to large proteins and protein fragments with inteins.225 227 Inteins are 100 800 residue polypeptide sequences found within proteins that are able to chemically excise
themselves and rejoin the parent protein with a peptide bond in a reaction catalyzed by an active thioester intermediate.225,228,229
This autocatalytic process is also referred to as intein-mediated protein splicing, and to date, more than 200 intein sequences have been identified in a wide range of protein families. Inteins are useful in applications such as protein synthesis, surface
immobilization, and conjugation with fluorescent probes or affinity handles.226,228,230 The chemistry of intein-mediated
processes is complex and quite diverse, and the interested reader is referred to other reviews for further details.225,226,228,229
As shown in Figure 11b, Rao’s group used intein chemistry to prepare QD-protein conjugates that functioned as bioluminescent protease sensors.231 A Renilla luciferase fusion protein with two in-line C-terminal peptide sequences was genetically engineered. The peptide sequences were a VPLSLTMG sequence recognized and cleaved by matrix metalloproteinase-7 (MMP-7), and a 198-residue GyrA intein sequence. Carboxyl-coated QDs were modified with adipic acid dihydrazide using carbodiimide coupling, and the thioester group of the intein underwent nucleophilic attack by the distal hydrazide of the QDs. This ultimately cleaved out the intein sequence and resulted in a QDluciferease conjugate linked through a protease substrate. In application, the addition of the luciferase substrate coelenterazine resulted in e cient bioluminescence resonance energy transfer (BRET) between the luciferase and the proximal QD. The addition of MMP-7 cleaved the bioconjugate linkage, disrupted BRET, and thus allowed monitoring of proteolytic
activity.231 Protein bioconjugates of liposomes and lipid bilayercoated silica NPs have also been prepared using split intein ligation or trans-splicing.232 In an elegant demonstration, two synthetic peptides—one modified with the C-terminal segment of a split DnaE intein, and the other modified with palmitoyl side chains for anchoring in a lipid bilayer—were joined by NCL and incorporated onto the lipid coated NPs. An eGFP fusion with the N-terminal segment of the split DnaE intein was then introduced and associated with the C-terminal segment resulting in excision of the intein to yield eGFP-NP conjugates.
Diazonium-Coupling to Tyrosine Side Chains. The reaction between a phenol and a diazonium salt to produce an azo compound is well-known in organic synthesis. In the case of proteins, diazonium salts can selectively react with the phenol and imidazole groups of tyrosine and histidine, respectively. The reaction to label the ortho position of tyrosine is illustrated in Figure 12a. Both tyrosine and histidine residues are typically lowabundance amino acids, found only sparingly in most proteins, and this may provide opportunities for site-specific labeling.
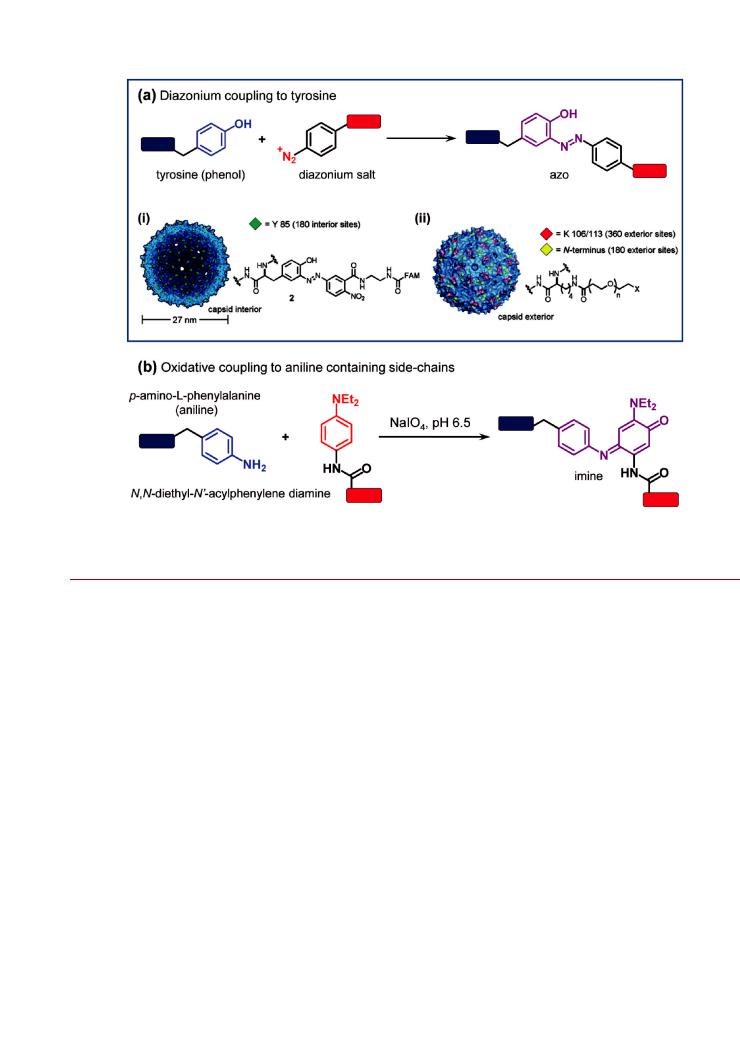
The Francis group has specifically modified tyrosine residues native to the interior surfaces of empty MS2 bacteriophage particles with nitro-phenyl diazonium salts.233 236 The nitrophenyl diazonium salts were linked to other functional groups that enabled the attachment of reporters such as organic dyes, MRI contrast agents, and radiolabels for positron emission tomography. As illustrated in Figure 12a, the diazonium reaction also allowed selective bioconjugation to the interior of the MS2 particles, while allowing lysine residues native to the exterior surface to be modified in orthogonal reactions. The latter included functionalization of the MS2 particles with succinimidyl esters of PEG and targeting ligands.235 Although a nitro-phenyl
diazonium salt derivative of a reporter may be prepared directly (e.g., fluorescein2,81,82), greater versatility has been achieved
through the use of nitro-phenyl diazonium salts that can introduce a bioorthogonal functional group at tyrosine residues. For example, the Francis group labeled MS2 tyrosine residues with aldehyde functionalized nitro-phenyl diazonium salts for subsequent oxime ligations,235 and developed a four-step heteroDiels Alder reaction that allowed further chemical modification.122 Diazonium coupling to tyrosine was used to introduce ketones on the surface of tobacco mosaic virus (TMV) particles for coupling to PEG through secondary oxime ligation.237 Similarly, Bruckman et al. have labeled TMV particles with alkyne-functionalized phenyl diazonium salts for subsequent CuAAC ligation with PEG, peptides, and dyes.238
Oxidative Coupling to Aniline-Containing Side Chains. The one-to-one oxidative coupling of aniline with N,N-diethyl-N0- acylphenylene diamine using sodium periodate was recently reported for the attachment of peptides to the surface of bacteriophage MS2.239 The reaction is shown in Figure 12b, and is both chemoselective and efficient at low reactant concentrations. A single residue of the unnatural amino acid p-amino- L-phenylalanine was incorporated into MS2 through amber suppression methods to facilitate this coupling. A peptide with a phenylene diamine modified N-terminus was then oxidatively coupled to the virus particles. The peptides selected for coupling allowed the conjugate to achieve specific delivery to neuroblastoma, breast cancer cell lines, and kidneys, and target the matrix metalloproteinase 2 and 9 enzymes. Oxidative coupling to aniline was also used to decorate the exterior surface of MS2 particles with zinc porphyrins for photocatalysis240 and a DNA aptamer for cellular delivery.241
841 |
dx.doi.org/10.1021/bc200065z |Bioconjugate Chem. 2011, 22, 825–858 |
Bioconjugate Chemistry |
|
REVIEW |
Figure 12. Coupling to protein/peptidyl side chains: (a) diazonium coupling to tyrosine residues; and (b) oxidative coupling to aniline containing side chains (e.g., p-amino-L-phenylalanine). The example for diazonium coupling to tyrosine shows (i) modification of tyrosine residues on the interior surface of an MS2 capsid with a fluorescent dye using a diazonium salt, and (ii) orthogonal coupling of PEG to exterior surface lysine residues using succinimidyl ester chemistry. Part of (a) adapted with permission from ref 216. Copyright 2007 American Chemical Society.
Self-Assembly. In contrast to the examples of the previous section, NP bioconjugate reactions based on self-assembly are not characterized by the formation of new covalent bonds. Rather, these reactions tend to be driven by comparatively weak coordination (or dative) bonding, wherein stability is dictated by equilibrium dissociation constants. Self-assembled NP bioconjugates are therefore sensitive to the concentrations of NP and biomolecule both during the preparation, and in subsequent application. However, through the use of moieties that selfassemble through multidentate coordination (e.g., polyhistidine vs monomeric histidine), the stability of self-assembled NPbioconjugates can be greatly improved and allow use at the low concentrations (e10 6 M) typical of biological applications. Compared to covalent methods, self-assembly methods are advantageous in that they generally offer rapid and facile bioconjugation without the need for additional reagents, and can often provide better control as a result.
Polyhistidine Coordination. The polyhistidine motif is wellknown for its role in the purification of proteins using immobilized metal ion affinity chromatography (IMAC) where it binds to Co2þ, Cu2þ, Ni2þ, Zn2þ and other divalent metal cations immobilized on solid supports through carboxylated chelates. These ions are also components of many different NP materials, including QDs and magnetic NPs. The imidazole ring of histidine, which coordinates divalent metal ions, can also interact with noble metals such as gold and silver. These properties suggest that polyhistidine can potentially be used to prepare
bioconjugates from a variety of inorganic NPs. Indeed, this has been confirmed experimentally with several different materials, although there is certainly further untapped potential.
Our research group pioneered the use of polyhistidine tags for self-assembly with QDs. As shown in Figure 13a, we found that histidine could coordinate to the Zn2þ ions at the inorganic surface of CdSe/ZnS QDs and drive the self-assembly of QDbioconjugates. The rapid and stable association of polyhistidine with the ZnS shell has proved highly advantageous for the
preparation of QD-bioconjugates with polyhistidine-tagged recombinant proteins242 and synthetic peptides.140,204,243 Disso-
ciation constants are typically in the range 10 1 102 nM and tend toward 1 nM in bulk solution, with equilibrium binding being reached within a few minutes at room temperature.244 As a consequence, polyhistidine self-assembly enables control over QD-bioconjugate valence through an approximately one-to-one correlation with stoichiometry. This correlation applies to the average valency, where the actual distribution of QD-bioconjugate valences is known to follow a Poisson distribution—especially at lower ratios.245 In addition, the polyhistidine motif o ers several intrinsic advantages for assembly to CdSe/ZnS QDs:
•The polyhistidine motif is bioorthogonal, since it is not normally found in natural proteins.
•The polyhistidine motif is routinely introduced to recombinant proteins to facilitate purification.
•The small size of polyhistidine motif does not disrupt native protein function.
842 |
dx.doi.org/10.1021/bc200065z |Bioconjugate Chem. 2011, 22, 825–858 |
Bioconjugate Chemistry |
|
REVIEW |
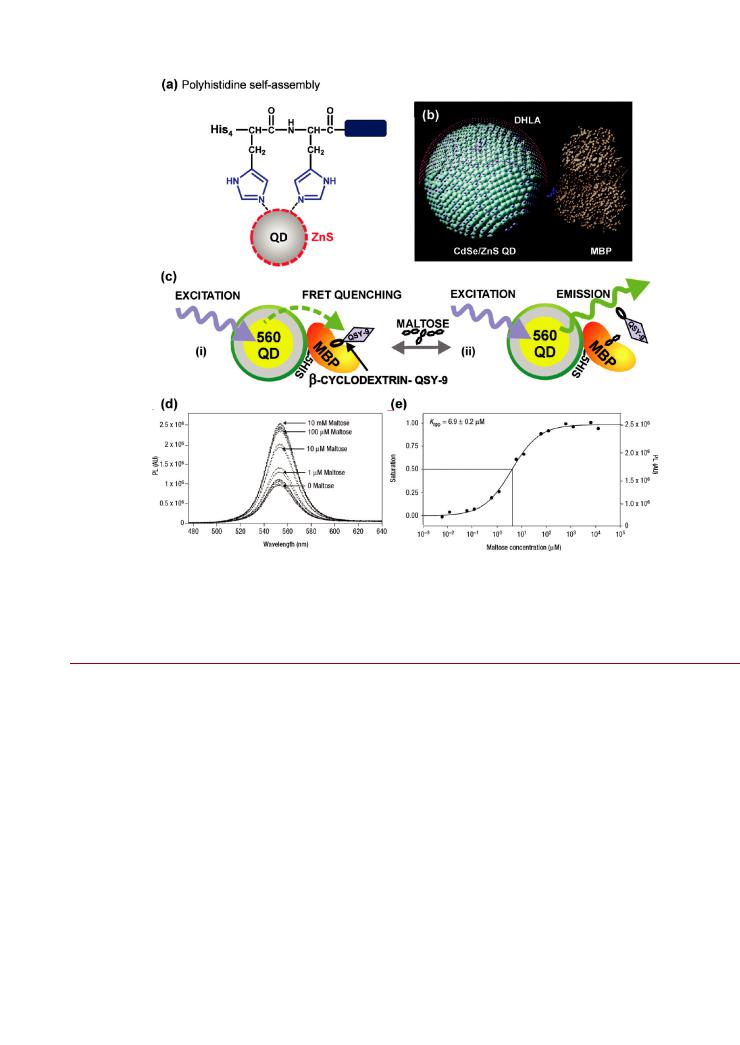
Figure 13. (a) Self-assembly of polyhistidine to the inorganic ZnS surface of a NP via direct coordination of the imidazole moieties. An example of the use of this bioconjugate chemistry is a FRET-based sensor for maltose using QD-His5-maltose binding protein (MBP) conjugates. An experimentally validated model of MBP assembled to a CdSe/ZnS QD is shown in (b). The polyhistidine tag is highlighted in blue. The sensing scheme (c) is based on
(i) binding of dark quencher (QSY-9)-labeled β-cyclodextrin by MBP, creating the proximity for quenching of the 560 nm emitting QD PL via FRET. The introduction of maltose (ii) displaces the quencher-labeled cyclodextrin and restores the QD PL in proportion to the maltose concentration. The QD PL recovery and a maltose binding isotherm are shown in (d) and (e), respectively. The image in (b) is reproduced from ref 246. The images in (c e) are reproduced from ref 256.
•The single point of attachment o ered by the polyhistidine motif eliminates undesirable cross-linking reactions and even enables control over biomolecular orientation.
Considering the latter point, a series of FRET experiments were used to determine the orientation of recombinant MBP assembled to a QD (Figure 13b). It was found that the polyhistidine tag interacted with the surface of the QD so as to orient the protein toward bulk solution.246 To further enhance control over orientation and spacing, we have exploited structural peptide motifs in combination with polyhistidines. These peptide motifs have included a rigid variable-length peptide based
on a repeating tyrosine-glutamic acid-histidine-lysine (YEHK) β-sheet,247,248 and the inclusion of a rigid helical linker-spacer
between an N-terminal polyhistidine tract and a C-terminal peptide suitable for use as proteolytic substrate.140 The rigid helical linker was based on R-amino isobutyric acid and alanine, with glycine hinges to separate its structure from the polyhistidine and proteolytic substrate.
Polyhistidine self-assembly with QDs has been extended to the preparation of QD-oligonucleotide conjugates through use of
modular “starter” peptide sequences with a hexahistidine tract at one terminus and a reactive group at the other. The latter have included pyridyl disulfide249 and iodacetyl groups250 that react with thiol-terminated oligonucleotides to form disulfide and thioether bonds, respectively. Thioether bonds are stable under most conditions; disulfides are labile under reducing conditions. Additional versatility is possible through polyhistidine starter peptides (Figure 10b) with a terminal benzaldehyde group that
enables hydrazone ligation to a peptide or oligonucleotide modified with a terminal HYNIC group.204,205 This bioorthogo-
nal chemistry is compatible with two di erent strategies: ligation with the polyhistidine starter peptide prior to assembly with QDs,204 or ligation after prior assembly of the polyhistidine starter peptide with QDs.205
The control provided by polyhistidine self-assembly has
greatly facilitated the development of QD-based cellular probes,243,251 and biosensors that use FRET (Figure 13c e)
or charge transfer for signal transduction.252 258 The interactions associated with the latter arise from conjugation with reporter-labeled biomolecules and are very sensitive to both
843 |
dx.doi.org/10.1021/bc200065z |Bioconjugate Chem. 2011, 22, 825–858 |

Bioconjugate Chemistry |
|
REVIEW |
the number of reporters per QD and the separation distance between reporter and QD, thus necessitating a high degree of control over the final bioconjugate architecture. In addition to CdSe/ZnS QDs, polyhistidine tagged recombinant enzymes have been assembled with CdS QDs.259 Magnetic Ni/NiO NPs and Au NPs have also been found to assemble with polyhistidine tagged proteins or peptides.260 262 A study of the assembly of polyhistidine tagged human acidic fibroblast growth factor with Au NPs found that the multichelate e ect of the histidine imidazole groups was able to displace the strong thiol Au NP interaction associated with a zwitterionic cy- steine-containing peptide coating.262 Dynamic measurements suggested that the fibroblast growth factor protein sampled a range of orientations at the Au NP surface before adopting a lowest energy state with attachment via the polyhistidine tag. While virus capsids have often been engineered with polyhistidine tags to facilitate purification, there is also the potential bioconjugation through polyhistidine. For example, heme groups have been conjugated to recombinant hepatitis B virus particles with N-terminal polyhistidine tags,263 and also to Qβ particles with C-terminal polyhistidine tags.264 Here, the assembly was driven by coordination to the iron center of heme.
Nickel(II) and Nitrilotriacetic Acid. Although direct polyhistidine coordination to the surface of NPs is a highly effective method for preparing controlled NP-bioconjugates, its application is limited to certain NP materials and thin coatings of solubilizing ligands that allow access to the inorganic surface of NPs. A more universal approach for the self-assembly of polyhistidine appended biomolecules to NPs is the use of nickel(II)- nitrilotriacetic acid (Ni2þ-NTA) modified NP coatings as shown in Figure 14a. This approach is borrowed from IMAC, where polyhistidine tags are known to bind to Ni2þ-NTA with dissociation constants on the order of Kd = 10 13 M.265 This affords many of the advantages and control associated with direct polyhistidine coordination while also being compatible with any NP material that displays a surface coating that can be chemically modified. The NTA-functionalization of a NP coating is facilitated by the availability of several NTA precursors and analogues. Many different NP materials have been modified
with NTA for binding polyhistidine tagged proteins, including QDs,265 268 Au NPs,269 CNTs,270 silica NPs,271,272 and mag-
netic NPs.273 The development of NTA-coated CNTs (Figure 14a) for controlled protein conjugation was motivated by the prior observation of protein denaturation on the hydrophobic surface of CNTs.270 This was minimized by the use of NTA for conjugation. Importantly, Susumu et al. demonstrated that the polyhistidine-Ni2þ-NTA interaction in QD conjugates remained stable when delivered to the cellular cytosol.268
The most common approach for introducing NTA has been to activate NP coatings toward a nucleophilic derivative of NTA. For example, QDs and magnetic NPs displaying carboxyl groups
have been activated with EDC/NHS toward NR,NR-bis(carboxy- methyl)-L-lysine—an amine-terminated derivative of NTA.266,267,273
Alternatively, succinic anhydride has been used to activate polyglycerol coated magnetic NPs toward the same lysine-NTA derivative,274 and glutaraldehyde has been used for the same purpose with amine terminated silica NPs.271 Silica NPs have also been directly modified with a silane derivative of NTA.272 Thiolated derivatives of NTA have been shown to self-assemble on Au NPs269 and QDs,275 or react with amine functionalized polymercoated QDs that had been activated with sulfosuccinimidyl- 4-(N-maleimidomethyl)cyclohexane-1-carboxylate (SMCC).265
Figure 14. Self-assembly of polyhistidine through coordination to Ni2þ supplemented (a) nitrilotriacetic acid (NTA) coated NPs or (b) carboxylate coated NPs. The example for (a) shows (i) an NTA modified CNT and (ii) Ni2þ supplementation with the introduction of a polyhistidine tagged protein (RC-His). The example for (b) shows
(i) the microinjection of carboxylate coated QDs into cells expressing polyhistidine tagged fluorescent protein (mCherry-His6) and (ii) fluorescence images showing the intracellular QD-protein assembly through FRET between the QD and mCherry. The white arrow indicates a control cell expressing mCherry but not injected with QDs. Part of (a) is adapted with permission from ref 270. Part of (b) is adapted with permission from ref 278. Copyright 2008 American Chemical SocietyCopyright 2010 American Chemical Society.
Using carbodiimide coupling, Au NPs coated with dihydrolipoic acid have been modified with Co2þ-NTA for self-assembly with polyhistidine tagged enzymes that largely retained their native activity.276 Similarly, magnetic NPs with Cu2þ-NTA have also been prepared.274
Chelating agents such as NTA rely, at least in part, on the ability of carboxyl groups in close proximity to simultaneously coordinate a metal ion. It appears that the density of carboxyl groups associated with NP coatings may also be su cient in this respect. Yao et al. demonstrated that carboxyl functionalized polymer-coated QDs were able to chelate Ni2þ directly, and thereby enable the controlled assembly of polyhistidine tagged Renilla luciferase enzyme for use in BRET-based sensing.277 Boeneman et al. found that this interaction is surprisingly robust and e cient and could be extended to an intracellular environment.278 Cells that expressed a polyhistidine tagged fluorescent protein (mCherry) were microinjected with Ni2þ-supplemented carboxyl polymer-coated QDs allowing the self-assembly process to occur in vivo (Figure 14b). This suggests suitability for similar in vivo bioconjugation of QDs with other target proteins.
Metallothionein Coordination. Metallothioneins (MTs) are low-molecular-weight proteins in which approximately one-third of the amino acid residues are cysteine. As a consequence, MTs have a high affinity for binding heavy metal ions. Similar to polyhistidine tags, MT tags that are appended to proteins can tightly bind the inorganic surface of QDs and Au NPs. The
844 |
dx.doi.org/10.1021/bc200065z |Bioconjugate Chem. 2011, 22, 825–858 |