

–
-
Реакции окисления в промышленном органическом и нефтехимическом синтезе
Окисление органических соединений занимает важное место в промышленном органическом и нефтехимическом синтезе. Окисляют парафиновые углеводороды, нафтены, арены, олефины, диены, спирты, альдегиды, алкины, меркаптаны, амины и др. органические соединения. В качестве окислителей используют O2, H2O2, ROOH, O3, N2O, неорганические окислители, включая Cl2, Br2, Cl2O и др.
Процессы окисления по разным признакам можно классифицировать следующим образом:
-
парциальное окисление;
-
глубокое окисление (обычно до СО2 и Н2О);
-
гомогенное газофазное окисление – обычно радикально-цепное автоокисление или инициированное окисление, процессы горения;
-
гомогенное гетерофазное (жидкофазное) окисление;
-
гетерогенно-каталитическое окисление.
Гомогенное жидкофазное окисление делится на радикально-цепное автоокисление (или инициированное окисление) и каталитическое окисление.
Приведем типичные реакции каталитического жидкофазного и гетерогенного окисления органических соединений.
-
Типичные окислители и реакции
Окисление кислородом
а) Радикально-цепное жидкофазное окисление алкилароматических соединений (катализ комплексами металлов)
![]()
![]()
б) “Мерокс”-процесс
![]()
в) “Вакер”-процесс (окисление олефинов)
![]()
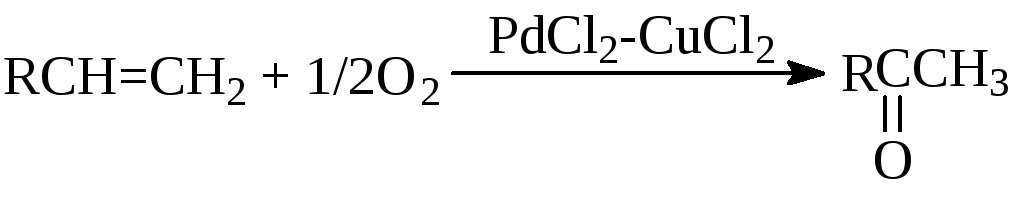
г) Реакция Моисеева (синтез винилацетата)
![]()
д) окислительное карбонилирование метанола
![]()
е) окислительная димеризация
Реакция Глязера-Залькинда:
![]()
Реакция Моритани-Фудживары:
![]()
ж) окислительное хлорирование
![]()
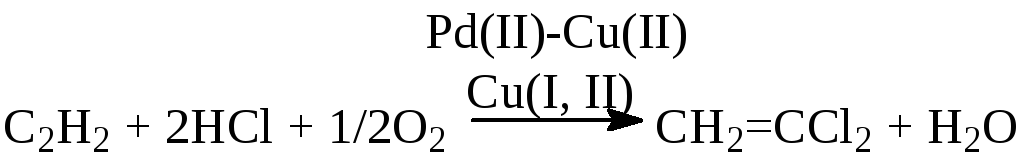
Окисление пероксидом водорода и гидропероксидами
а) Реакция Прилежаева

б) Эпоксидирование олефинов
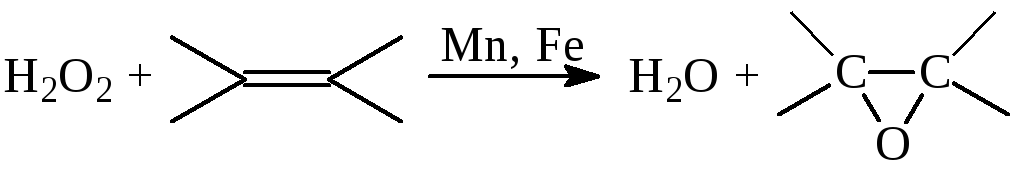
в) Окисление аренов и фенолов
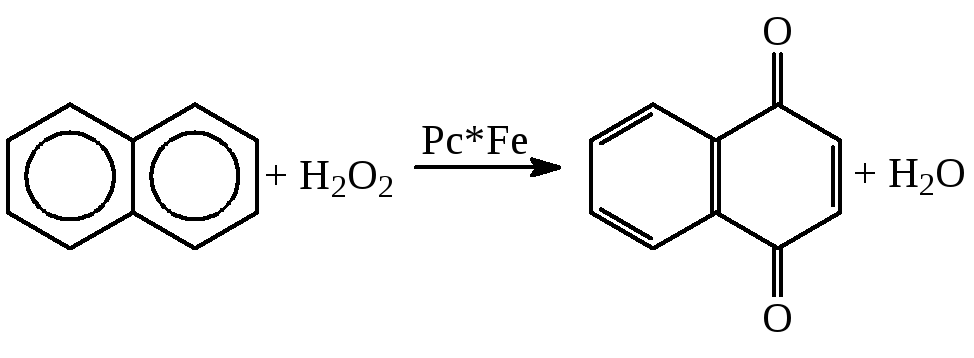
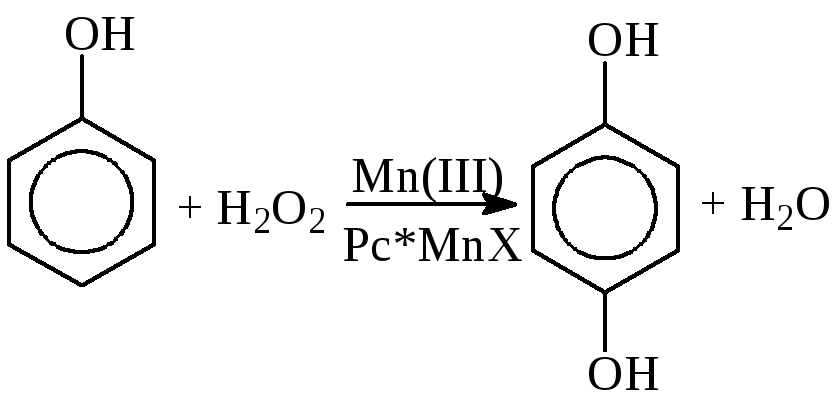
Pc* – замещенные фталоцианины
г) “Халкон”-процесс
Окисление О2 в гетерогенном катализе
а) окисление спиртов
![]()
![]()
б) окисление ароматических соединений
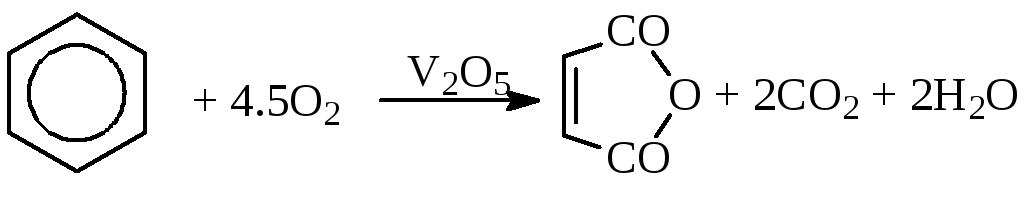
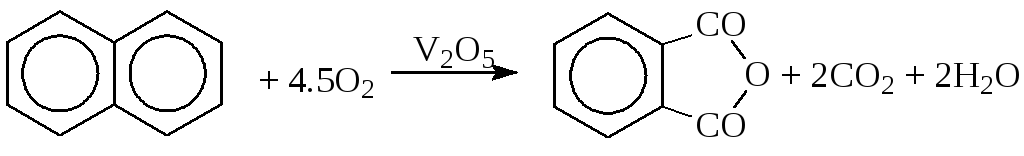
в) окисление алканов (окислительное дегидрирование)
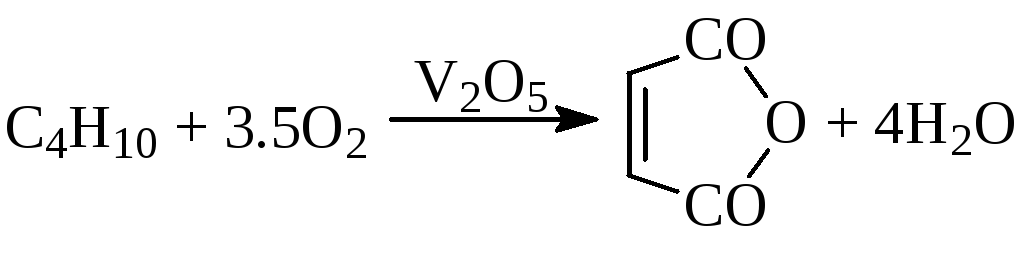
г) окисление олефинов
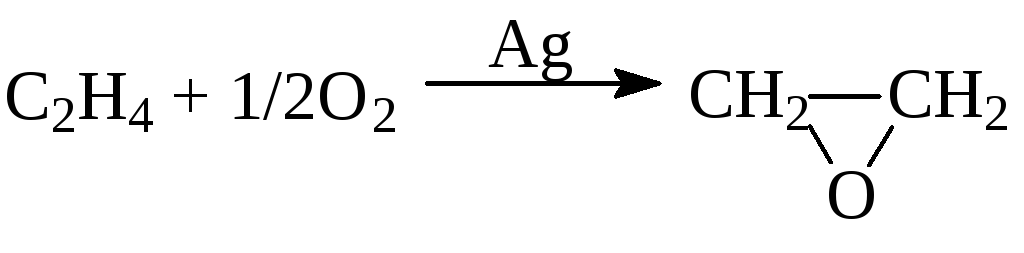
![]()
д) окислительный аммонолиз парафинов и олефинов
![]()
![]()
е) реакция Моисеева в паровой фазе
![]()
ж) синтез аллилацетата
![]()
з) окислительная димеризация метана
![]()
и) окислительное хлорирование этилена
![]()
Появились и новые окислители, например, закись азота N2O. Бензол окисляется этим окислителем на цеолитах ZSM-5, содержащих железо, при 350 – 400оС. Селективность ~100%, конверсия 8 – 13%.
![]()
Недавно (в 2002 г) установлено (Г.И.Панов), что N2O в жидкой фазе без катализатора при давлении 10 атм и температурах 140 – 250оС окисляет олефины до кетонов с селективностью > 98%.
-
Некоторые процессы, механизмы и кинетические модели
“Вакер”-процесс. Реакция окисления олефинов до карбонильных соединений была открыта практически одновременно в Германии (группа доктора Юргена Смидта в фирме “Consortium für Electrochemie”) и И.И.Моисеевым, М.Н.Варгафтиком и Я.К.Сыркиным в СССР (МИТХТ им. М.В.Ломоносова) в 1957 – 1959 гг. Реакция протекает в воде или водно-органических растворах комплексов Pd(II) и Cu(II) при атмосферном давлении и температурах 70 – 95оС, например, синтез ацетальдегида:
![]() (1)
(1)
Реакция (1) вызвала интерес у промышленных фирм, и уже в 1962 году фирма “Wacker Chemie” построила производство альдегида по этой реакции. В промышленных условиях используют давление 10 – 13 атм и температуру 110 – 120оС. Процесс (1) складывается из трех макростадий (2 – 4):
![]() (2)
(2)
![]() (3)
(3)
![]() (4)
(4)
Таким образом, PdCl2 катализирует окисление этилена окислителем CuCl2 (стадии (2) и (3)), а CuCl2 катализирует окисление Pd0 кислородом (стадии 3, 4). Система PdCl2-CuCl2 является полифункциональным катализатором брутто-процесса (1). Интересно, что молекула воды также катализирует брутто-реакцию и является непременным участником процесса в этой каталитической системе. Поскольку скорость окисления Cu(I) кислородом достаточно велика, стационарность процесса обеспечивается равенством скоростей реакций (2) и (3). В условиях промышленного процесса скорость реакции (3) обеспечивает отсутствие Pd0 в форме металлической фазы, и скорость реакции (1) в определенных пределах не зависит от [CuCl2]. Вместо CuCl2 можно использовать другие промежуточные окислители, например, п-бензохинон, концентрация которого при определенном избытке также не влияет на скорость образования ацетальдегида. Эту систему и использовали для построения кинетической модели и изучения механизма реакции. Очевидно, таким образом, что главные события, приводящие к очень интересному превращению этилена с участием H2O, происходят в реакции (2).
Кинетическое уравнение для реакции (2) в присутствии п-бензохинона (Q) было получено в закрытой системе без газовой фазы (И.И.Моисеев и др.) и по поглощению этилена в двухфазном реакторе полного смешения волюмометрическим методом (П.Генри). В области концентраций PdCl2 до 0.02 М при постоянной ионной силе (I = 1 – 3) в системе NaCl – LiClO4 – HCl – HClO4 = Const Pd(II) находится преимущественно в форме PdCl42– , и закомплексованность Pd(II) этиленом не существенна. Скорость реакции (2) или реакции (5)
![]() (5)
(5)
описывается уравнением (6)
![]() (6)
(6)
Из уравнения (6) следует, что процесс протекает с лимитирующей стадией и что в стадиях до лимитирующей выделяются ион Н+ и два иона Cl– при взаимодействии PdCl42– и C2H4. Для выяснения вопроса о том, из какой частицы выделяется Н+, провели опыты с меченым этиленом (C2D4) в H2O. Оказалось, что ацетальдегид содержит 4 атома D (CD3CDO) и, таким образом, Н+ может выделяться только из молекулы H2O. Схема механизма, соответствующая уравнению (6) и подтвержденная независимым исследованием равновесий в этой системе, включает стадии
![]() (7)
(7)
![]() (8)
(8)
![]() (9)
(9)
![]() (10)
(10)
![]() (11)
(11)
![]() (12)
(12)
![]() (13)
(13)
Механизм лимитирующей стадии (10) и механизм стадии (11) до сих пор являются предметом дискуссий.
Для расчетов промышленного реактора в случае системы PdCl2-CuCl2 в условиях постоянной концентрации HCl по длине трубчатого реактора (труба в трубе) используют несколько измененное уравнение, найденное экспериментально на основе уравнения (6). Скорость накопления ацетальдегида (САА) или исчезновения этилена вдоль трубы длиной l описывают уравнением
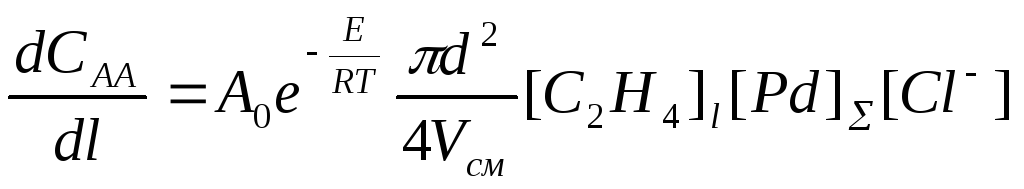 . (14)
. (14)
где d – внутренний диаметр трубы, м; Vсм – объем смеси этилена и раствора, поступающего в трубу, Vсм 0.5 м3/сек; [С2Н4]l – концентрация этилена вдоль трубы в молях на м3, рассчитываемая по найденной зависимости [С2Н4]l = f (T, P)
![]() ,
,
где
![]() ,
см
– плотность смеси в кг/м3;
,
см
– плотность смеси в кг/м3;
![]() ,
P0 – общее
давление смеси, P
– понижение давления по длине трубы; l
– длина трубы.
,
P0 – общее
давление смеси, P
– понижение давления по длине трубы; l
– длина трубы.
Синтез винилацетата (реакция Моисеева). Реакция окислительной этерификации или окислительного ацетоксилирования олефинов
![]() (15)
(15)
была открыта в МИТХТ им. Ломоносова в 1960 г. Реакция осуществляется в растворах солей PdCl2-CuCl2 и Cu(OAc)2 в уксусной кислоте в присутствии NaOAc. Температура процесса 110 – 130оС и давление 3.0 – 4.0 МПа. Селективность по этилену – 83%. Кинетическое уравнение получено Моисеевым и Беловым в системе, не содержащей CuCl2 (16)
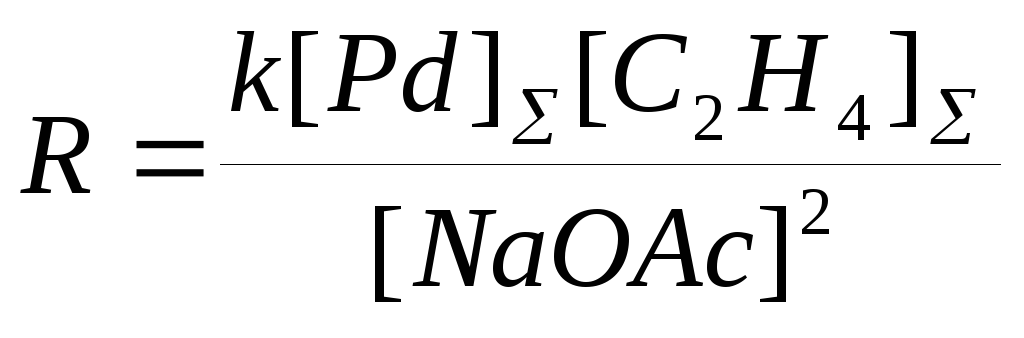 (16)
(16)
в предположении, что в условиях квадратичного торможения ацетатом натрия весь Pd(II) находится в форме комплекса Na2Pd(OAc)4. В работе П.Генри приведена другая форма уравнения (16) в предположении, что активной формой Pd(II) является димер Na2Pd2(OAc)6, концентрация которого проходит через максимум по [NaOAc]
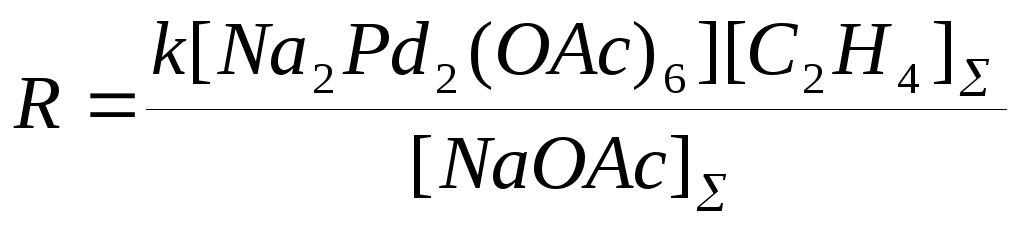 (17)
(17)
Процесс синтеза винилацетата по реакции (15) протекает в рамках механизма, аналогичного “Вакер”-процессу. Предполагается превращение -комплекса Pd(II) в -палладийорганическое соединение под действием OAc– из раствора, а распад полученного интермедиата включает стадию -элиминирования ~PdH
![]() , (18)
, (18)
где [Pd] – мономерный или димерный комплекс Pd(II). Окислением H-[Pd] и заканчивается каталитический цикл.
Фирмы Hoechst и др. разработали для реакции (15) гетерогенный катализатор, содержащий соли Pd(II), Au(III) и KOAc на Al2O3. Процесс протекает при 175 – 200 оС и давлении 0.5 – 1.0 МПа с высокой селективностью: 94% по этилену и 98% по уксусной кислоте. Состояние Pd(II) в условиях процесса и роль соединений золота пока не ясны.
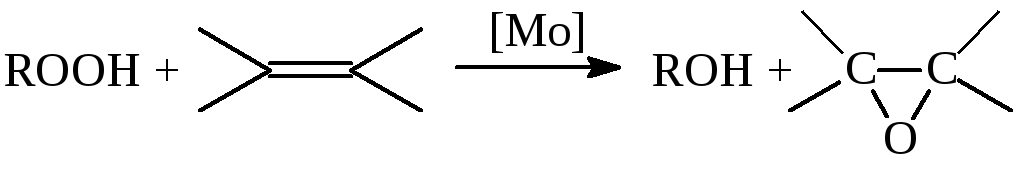
Халкон-процесс. Эпоксидирование олефинов гидропероксидами осуществляется в промышленном варианте в растворах комплексов Mo(VI). В качестве ROOH используют 2-этилфенилгидропероксид (гидропероксид этилбензола, ГПЭБ), гидропероксид кумила (ГПК) и третбутилгидропероксид (ТБГП). В случае ГПЭБ сопряженно с пропиленоксидом получают стирол:
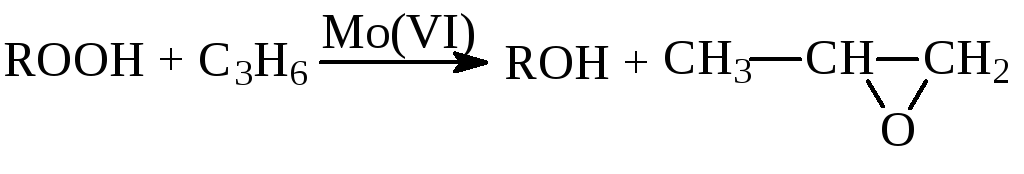 (18)
(18)
![]() (19)
(19)
Скорость реакции (18) описывается уравнением (20)
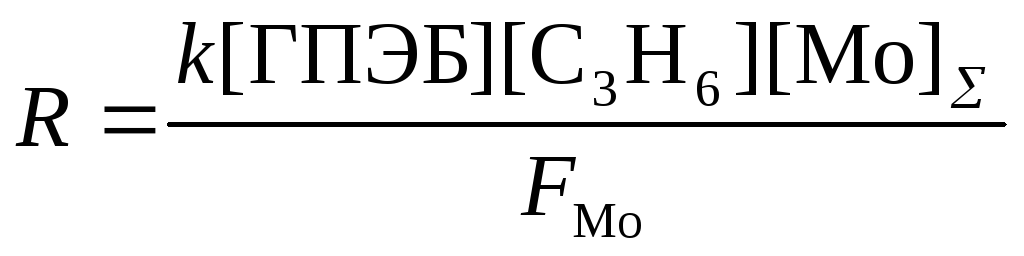 (20)
(20)
где FMo = 1 + KГПЭБ[ГПЭБ] + KМФК[МФК] + KОП[ОП] + KH2O[H2O] есть закомплексованность катализатора, МФК – метилфенилкарбинол, ОП – пропиленоксид. Ki – константы равновесия образования соответствующих комплексов Mo. Как видно из уравнения (20), процесс протекает с лимитирующей стадией, переходное состояние которой включает ГПЭБ, Mo(VI) и пропилен. Показано, что активным катализатором является пропиленгликолятный комплекс Mo(VI), реакция которого с ГПЭБ и C3H6 приводит к ОП.
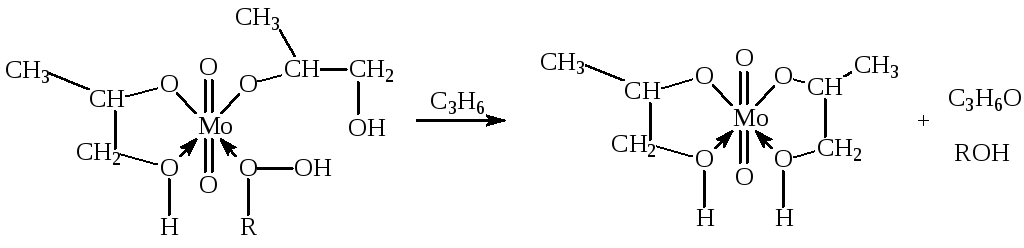
Мерокс-процесс. Реакция окислительной димеризации меркаптанов
![]() (21)
(21)
является основой процесса демеркаптанизации природного газа, попутных газов и нефтяных фракций, разработанного фирмой UOP. В водных растворах комплексов Co(II) (Pc*Co, Pc* – замещенный сульфофталоцианин) в присутствии NaOH происходит процесс образования радикалов RS·, димеризация которых дает RS-SR.
![]()
![]()
![]()
![]()
![]()
Образующиеся Co(III) и H2O2 также окисляют RSH до RS-SR, и в результате получается реакция (21). Нерастворимый дисульфид отделяется от воды, а водный раствор NaOH с катализатором направляется на экстракцию RSH из газа и нефти.
Окислительная димеризация алкинов (реакция Глязера-Залькинда) занимает важное место в синтетической химии.
![]() (22)
(22)
В этой реакции, в отличие от Вакер-процесса, оба компонента каталитической системы Cu(I) и Cu(II) принимают участие в образовании продукта, а О2 (или другой окислитель, Q, Fe(CN)63– и т.д.) регенерирует необходимую для реакции форму Cu(II). Дегидроконденсацию алкинов можно провести в электрохимической системе (в анодной камере электролизера), например, по реакции
![]()
При использовании в качестве окислителя Cu(OAc)2 в Ру реакция является автокаталитической. В системе CuCl-CuCl2-LiCl-H2O при большом избытке LiCl (т.е. при постоянной концентрации Cl–) скорость димеризации метилацетилена описывается уравнением
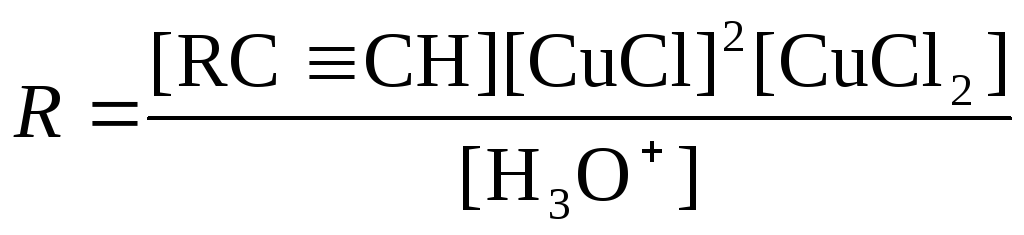 , (23)
, (23)
свидетельствующем о наличии лимитирующей стадии и аниона RCC– в переходном состоянии лимитирующей стадии
[(RCC–)·2Cu(I)·Cu(II)]≠
Таким образом, продукт превращения интермедиата RCCCu·CuCl в реакции с CuCl2 (Х1) и является интермедиатом, участвующим в образовании диалкина. Предполагается следующая схема реакции:
![]() (24)
(24)
![]() (25)
(25)
![]() (26)
(26)
![]() (27)
(27)
Образование радикала RCC· в стадии (26) (с его последующей димеризацией) не проходит по термохимическим соображениям. В Мерокс-процесе стадия с участием RS· возможна. Похожая на (22) реакция димеризации HCN также осуществляется в растворах Cu(I)-Cu(II)
![]() (28)
(28)
Гидролиз дициана дает оксамид NH2COCONH2 – очень ценное удобрение.
Синтез оксида этилена. Этиленоксид (ЭО) получают по реакции (29)
![]() (29)
(29)
на серебряных катализаторах 15% Ag/-Al2O3 при 240 – 270оС и давлении 3МПа. При конверсии этилена < 10% селективность 80 – 85%. Побочная реакция – глубокое окисление этилена до СО2. Селективность процесса повышают добавками Cl (NaCl) в катализатор или добавками дихлорэтана в сырье в количестве 2 – 10 ppm. СО2 образуется из С2Н4 и при окислении ЭО, поэтому химизм процесса определяется совокупностью параллельно-последовательных реакций
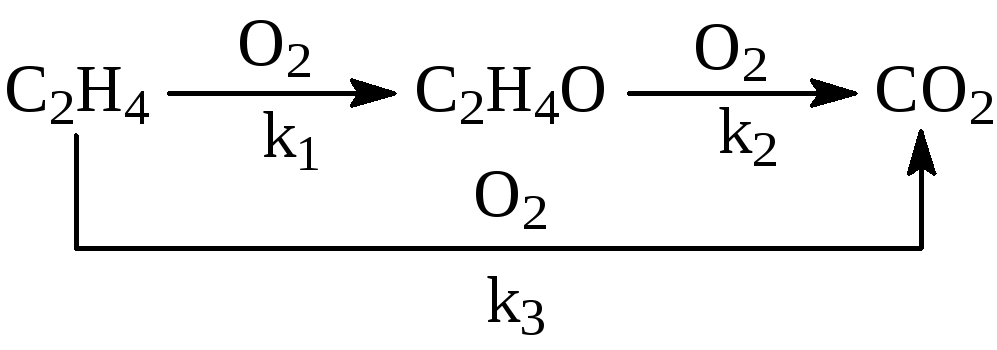
Обсуждаются различные гипотезы о механизме процесса, предполагающие образование СО2 на тех же центрах ZO2, на которых образуется ЭО, или участие разных центров в образовании ЭО (ZO2) и СО2 (ZO). Скорость расходования О2 в области PC2H4 > 0.9 атм на промотированном хлором катализаторе описывается уравнением первого порядка по РО2 (лимитирует адсорбция О2). При РО2 > 0.5 атм и PC2H4 ≤ 0.02
![]() (30)
(30)
Для очень простой схемы
![]()
![]()
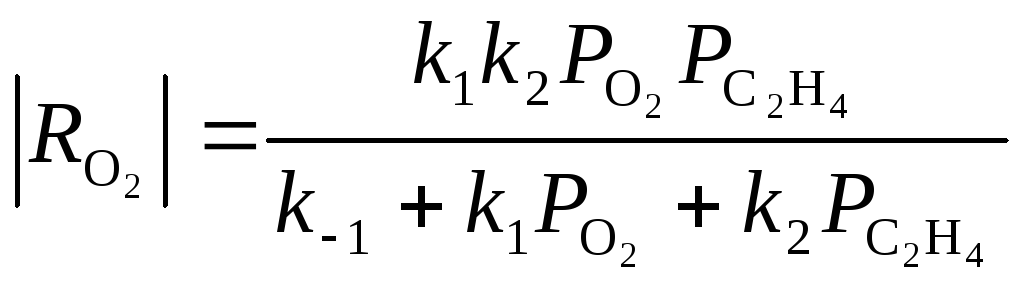 (31)
(31)
Из уравнения (31) получаются оба частных случая. Процесс тормозится ЭО и СО2, поэтому, например, при PC2H4 > 0.9 атм в условиях первого порядка по РО2
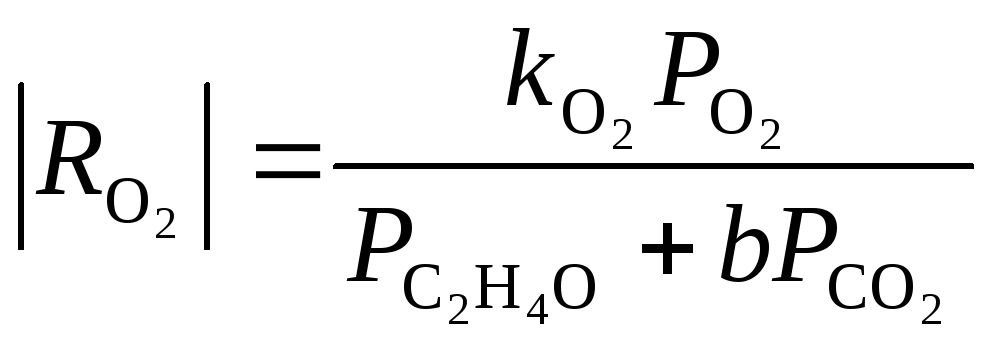 (32)
(32)
Если поверхностные соединения серебра и кислорода представить в виде химических соединений, то центрам Z, ZO2 и ZO можно сопоставить Ag2O, Ag2O3 и Ag2O2, соответственно. Имеются и другие представления об адсорбированных на поверхности серебра формах кислорода, в том числе и об участии в реакции атомов кислорода (или О–), находящихся в приповерхностном слое.
Окисление спиртов. Окисление (или окислительное дегидрирование) спиртов на металлических и окисных катализаторах до альдегидов и кетонов является важным промышленным процессом.
Рассмотрим подробнее процесс окисления метанола до формальдегида
![]() (33)
(33)
В промышленности реализованы два варианты процесса окисления:
-
на оксидах MoO3-Fe2O3 (и др. оксидных катализаторах) процесс протекает в кинетической области при 300 – 350оС и 15-кратном избытке воздуха по отношению к метанолу. При этом достигаются 100% превращение спирта, высокая селективность и синтез безметанольного формальдегида, необходимого для процессов его полимеризации.
-
На серебряных катализаторах (мелкокристаллическое серебро, Ag/пемза, Ag/-Al2O3 и др.) процесс протекает в адиабатическом режиме в тонком слое катализатора (8 – 10 см) во внешнедиффузионной области. Количество подаваемого кислорода ~0.9 от стехиометрии.
И основная реакция (33), и побочная реакция (34)
![]() (34)
(34)
– экзотермические процессы. Эндотермический процесс дегидрирования (35), который имеет место в условиях процесса
![]() (35)
(35)
не компенсируют большого количества выделяющегося тепла. Поэтому при низких температурах (220 – 250оС) процесс протекает в кинетическом режиме, однако при больших нагрузках по спирту и небольшом количестве воздуха процесс не удерживается в изотермическом режиме, и начинается быстрый подъем температуры, обусловленный плохим отводом тепла и повышением температуры зерна катализатора Тз. Повышение Тз вызывает экспоненциальный рост скорости, рост количества выделяющегося тепла qподв (ккал/(л·час)) и еще больший рост Тз, который останавливается в новом стационарном состоянии при высоком градиенте Тз – Tf (Tf – температура газа), обеспечивающем равенство отводимого и подводимого тепла qподв qотв. Таким образом:
-
в области низких температур Тз Tf, qподв qотв, процесс протекает в кинетической области (область i);
-
при повышении Тз возникает неустойчивый режим (область n);
-
при Тз > Tf режим адиабатический (qподв qотв), внешнедиффузионная область, режим “зажигания”, работает тонкий слой катализатора (область k).
Температуру адиабатического разогрева можно оценить по уравнениям
![]() ;
;
![]() ,
,
где – объемный коэффициент теплоотдачи (кал/(л·час·гр)), Q – количество выделяемого тепла (кал/моль), Cf – скорость реакции в диффузионном режиме, – коэффициент скорости диффузии, Cf – концентрация спирта в потоке.
При равенстве qподв = qотв,
![]() , (36)
, (36)
где – теплоемкость, кал/(л·гр).
Режим зажигания устанавливается при
![]() ,
где
,
где
![]() ,
,
Е – наблюдаемая энергия активации процесса.
В режиме диффузионного “зажигания” Tf = 650 – 700оС, Тз = 900 – 1000оС, но при малых временах контакта селективность процесса достигает 95% при 90% конверсии метанола. Полученный в результате абсорбции водой раствор 40% формальдегида (формалин) можно использовать как товарный продукт.
Окислительное хлорирование этилена до дихлорэтана. Процесс синтеза дихлорэтана (ДХЭ) по реакции (37)
![]() (37)
(37)
протекает в области 325 – 525оС (лучше 350 – 400оС) на меднохлоридных катализаторах CuCl-KCl/SiO2 или CuCl2/-Al2O3 практически при 100% конверсии HCl с выходом ДХЭ по этилену ~ 96%. Дихлорэтан образуется на поверхности катализатора без участия свободного Cl2. Механизм реакции изучен весьма детально. Схема механизма приведена ниже для второго катализатора.
![]()
![]()
![]()
![]()
![]()
![]()
Если вектор стехиометрических чисел стадий маршрута равен |2 2 2 2 1 1|, получим итоговое уравнение (37). Скорость образования ДХЭ описывается уравнением (38) с учетом 2-х медленных стадий (3) и (5):
![]() , (38)
, (38)
где
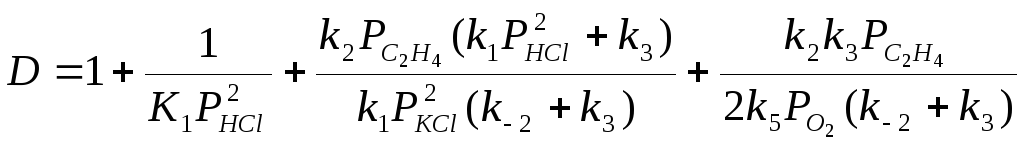 .
.
При PHCl 2 кПа реализуется нулевой порядок по PHCl, и при определенных соотношениях констант уравнение (38) преобразуется к виду (39)
 (39)
(39)