

макет_зуб
.pdfЛІТЕРАТУРА
1.Накамото, К. ИК спектры и спектры КР неорганических и координационных соединений / Накамото К. – М. : Мир, 1991.
2.Nakamoto, K. Infrared and Raman Spectra of Inorganic and Coordination Compounds / Nakamoto K. – Hoboken, New Jersey : John Wiley&Sons, Inc. 2009.
Допоміжна
3.Григорьев, А.И. Введение в колебательную спектроскопию неорганических соединений / Григорьев А.И. – М. : Изд-во Моск. ун-та, 1977.
4.Жижин, Г.Н. и др. Оптические колебательные спектры кристаллов / Жижин Г.Н., Маврин Б.Н., Шабанов В.Ф. – М. :
Наука, 1994.
5.Khavryuchenko, V. Computation Vibration Spectroscopy as a Tool for Investigation of Complicated Systems / Khavryuchenko V.
//Euras.Chem.Tech. J. – 2004. – Vol. 6. – P. 157.
83

Розділ 3 ВИКОРИСТАННЯ КОНЦЕПЦІЇ ГРУПОВИХ (ХАРАКТЕРИСТИЧНИХ) КОЛИВАНЬ ДЛЯ АНАЛІЗУ ІЧ СПЕКТРІВ
Інтерпретація коливальних спектрів молекул і іонів, які мають досить високу симетрію, як було показано вище, неможлива без детального аналізу форми та симетрії їх коливань. Симетрія більшості складних молекул є низькою і тому їх спектри не мають вироджених чи заборонених за симетрією коливань. Строге вирішення обернено-коливальної задачі для складних молекул є справою непростою та потребує спеціальних знань і тому хіміками використовується досить рідко. У переважній більшості досліджень для інтерпретації ІЧ спектрів складних речовин використовують більш прості методи, які в першу чергу основані на понятті про характеристичність смуг поглинання.
Як уже зазначалось у розд. 1, якщо для нормального коливання складної молекули всі коефіцієнти форми Ci, за винятком одного, є близькими до нуля, то таке коливання є характеристичним (груповим) для цього зв'язку чи зазначеної групи атомів, тобто воно локалізоване практично на окремій функціональній групі і саме вона визначає форму та частоту цього коливання. Таке коливання вважається характеристичним за формі і за частотою.
84
Узагалі поняття характеристичності виникло в ІЧ спектроскопії емпірично на основі аналізу великої кількості експериментальних даних. Було встановлено, що деяким функціональним хімічним групам відповідають смуги поглинання у вузькій частині спектра незалежно від природи молекули, у складі якої вони містяться. Вплив інших атомів і зв'язків молекули на положення й інтенсивність цих смуг поглинання є незначним. Такі характеристичні коливання широко використовують для якісного, а іноді й кількісного аналізу складних речовин, у першу чергу органічних сполук і координаційних сполук з органічними лігандами. Крім того, якщо коливання є характеристичними, то їх частоти (точніше хвильові числа) можна використовувати замість силових сталих для аналізу природи хімічного зв'язку при тих чи інших змінах у складі молекул (уведення замісників, координації іоном металу тощо). Так, наприклад, зменшення частоти валентних коливань ν(СО) у певному ряду речовин може свідчити про зменшення порядку зв'язку С–О в цьому ряду.
Які ж типи коливань можна вважати характеристичними за частотою? У першу чергу, це валентні коливання за участі легких атомів гідрогену, тобто ν(Е–Н), при яких легкий атом гідрогену коливається відносно більш важкого атома, як, наприклад, при валентних коливаннях С–Н, N–H, О–Н тощо. У цьому випадку коливальний рух сконцентровано на більш легкому атомі, а атом іншого елемента практично не зміщується. Тому це коливання зазнає лише незначного впливу решти частини молекули і є характеристичним за формою та частотою. Характеристичними є також коливання за участі, наприклад, атомів галогенів (C– Cl, C–Br), які суттєво більші за масою від інших атомів у молекулі. Відносно незалежними можуть бути також валентні коливання груп із ізольованими (не спряженими) кратними зв'язками, наприклад Р=О, S=O, С=О, С=С, С≡N тощо. Характеристичні частоти майже завжди легко виявити, оскільки вони потрапляють у відносно вузькі частотні інтервали спектрів, і тому їх широко використовують в ІЧ спектроскопії для аналізу якісного складу речовин.
Деякі коливання можуть бути нехарактеристичними за формою, але бути наближено характеристичними за частотою. Так,
85
наприклад, як бачимо з табл. 2.4, коливання ν3 нітрат іона за формою є валентно-деформаційним коливанням, (50 % νN–O + 50 % δONO), а за енергетичним вкладом, тобто за частотою, його можна вважати асиметричним валентним коливанням, νasNO (95 %
νN–O + 5 % δONO). Таким чином, це коливання є нехаректеристичним за формою, але наближено характеристичним за частотою.
Якщо атоми мають близьку масу та зв'язані між собою одинарними зв'язками, то вони, як правило, не є характеристичними, оскільки коливання одного зв'язка викликає синхронну зміну довжин сусідніх зв'язків і кутів (іноді говорять, що відбувається змішування коливань). Такі коливання розташовані переважно в області 1300–800 см–1. Проте навіть у цій області ІЧ спектра є деякі смуги поглинання, які можна легко ідентифікувати за інтенсивністю. Це коливання полярних зв'язків, таких як C–O, N–O, P–O і їм подібних, при коливанні яких значно змінюються дипольні моменти, і тому відповідні смуги поглинання в ІЧ спектрах мають дуже велику інтенсивність.
Область 1300–800 см–1 іноді називають областю "відбитків пальців" (англ. "finger prints"). Таку назву ця область одержала завдяки тому, що кількість, частота й інтенсивність смуг поглинання, які містяться в ній, є індивідуальною характеристикою даної речовини (її ІЧ паспортом). Це дозволяє за ІЧ спектром ідентифікувати навіть близькі за будовою сполуки, наприклад геометричні ізомери, які розрізнити іншими методами значно важче.
Із появою Фур'є ІЧ спектрометрів з'явилися широкі можливості для проведення швидкої ідентифікації навіть дуже складних речовин за їх ІЧ спектрами. Для цього записують спектр невідомої речовини та порівнюють його зі спектрами з комп'ютерної бази даних, яка на сьогодні містить ІЧ спектри тисяч різних речовин. При цьому, у першу чергу, важливим є збіг смуг поглинання саме в області "відбитків пальців". Такий експрес-аналіз займає декілька хвилин і часто дозволяє не тільки ідентифікувати речовину, але і визначити, яким методом, і в якій лабораторії її було одержано.
86
3.1. ФАКТОРИ, ЯКІ ВПЛИВАЮТЬ НА ЗНАЧЕННЯ ХАРАКТЕРИСТИЧНИХ (ГРУПОВИХ) ЧАСТОТ
Частота коливання відповідно до формули (6) залежить від значення силової сталої, яка є фундаментальною характеристикою зв'язку поряд з енергією та довжиною зв'язку. Силові сталі, у свою чергу, значно залежать від порядку зв'язку: чим більший порядок зв'язку, тим більша його силова стала і тим вища частота коливання відповідного зв'язку. Так, частоти коливання потрійних зв'язків за участі атомів карбону, нітрогену й оксигену, які мають близькі значення атомних мас (а це переважна більшість органічних сполук), розташовані в області 2300–2000 см–1, подвійних зв'язків – в області 1800–1500 см–1, а одинарних – нижче 1500 см–1. Ці спектральні області іноді називають областями коливання одинарних, подвійних чи потрійних зв'язків. Проте, якщо в утворенні зв'язків беруть участь атоми, які мають значно більшу або меншу маси, то частоти коливання матимуть зовсім інші значення, наприклад частота валентного коливання ν(С=S) розміщується в області 800–700 см–1, а одинарних зв'язків N–Н чи О–Н в області 3600–3200 см–1.
В ІЧ спектрах іноді вдається зареєструвати і валентні коливання водневих зв'язків, ν(О…Н), але вони перебувають в області нижче 400 см–1, оскільки силові сталі для таких зв'язків мають дуже мале значення. Наприклад, частота коливання водневого зв'язку Н...О в мурашиній кислоті є рівною 248 см–1, тоді як валентні коливання О–Н містять близько 3600 см–1.
Силові сталі деформаційних коливань значно менші від силових сталих валентних коливань, тому частоти валентних коливань завжди мають більші значення порівняно з частотами деформаційних коливань за участі тих самих атомів. Наприклад, якщо валентні коливання зв'язків N–Н чи О–Н розташовані в області 3600 –3200 см–1, то частоти деформаційних коливань δ(Н2О) і δ(NН2) мають значення близьке до 1600 см–1. Подібну залежність між валентними й деформаційними коливаннями добре також видно з таблиць, у яких наведені частоти коливань неорганічних молекул та іонів (табл. 2.5–2.8).
87
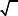
У розд. 1 було показано, що частота коливання залежить від зведеної маси атомів (1.3). Ця залежність наближено виконується і для характеристичних коливань у багатоатомних молекулах: частота коливання є обернено пропорційною зведеній масі атомів, які беруть участь у цьому коливанні. Валентні коливання за участі атомів гідрогену розташовані вище 2500 см–1, коливання зв'язків за участі атомів другого періоду (карбон, нітроген, оксиген, фтор) – в області 1800–1000 см–1, коливання за участі атомів третього періоду – в області 1000–400 см–1. Звичайно наведені числа є наближеними, але досить чітко відображають загальну тенденцію залежності частот коливання від маси атомів. Слід зауважити, що, на жаль, коливання багатьох неорганічних сполук важких металів: коливання метал – ліганд у координаційних сполуках, коливання метал – метал у кластерах, як правило, потрапляють у далеку ІЧ область (400–200 см–1), яка розміщується за межами робочої зони більшості ІЧ спектрометрів. Це ж стосується і водневих зв'язків у органічних молекулах (див. вище).
Оскільки частоти коливань залежать від маси атомів, заміщення останніх на ізотопи приводить до зміни частот при незмінній геометрії молекули та силових сталих. Такі зміни у спектрах називають ізотопним ефектом. Різниця частот ізотопозаміщених молекул залежить від співвідношення мас ізотопів mi/m, де mi – маса більш важкого ізотопа. Так, наприклад, при заміні атомів гідрогену на дейтерій смуги поглинання зсуваються в низькочастотну область приблизно в 1,3–1,5 разів
(у 2 разів), причому зсув смуг поглинання мало залежить від типу коливання. Наприклад, νas(ОН)/νas(OD) = 3450/2790 = 1,24; νs(ОН)/νs(OD) = 3210/2670 = 1,2; δ(HOH)/δ(DOD) = 1630/1180 = 1,38.
При заміні ізотопа 16О на 18О частота відповідного коливання зменшується приблизно на 40 см–1. Використання ізотопного ефекту дає додаткову інформацію для інтерпретації коливальних спектрів. На рис. 3.1 наведено ІЧ спектри (СН3)2SО і (CD3)2SO. Смуги поглинання, які зазнають значного низькочастотного зсуву, належать до валентних і деформаційних коливань СН3 груп, а смуга поглинання при 1060 см–1, яка не змінює свого положення, належить до валентного коливання ν(S=O).
88
1
2
4000 3600 3200 2800 2400 2000 1600 1200 800 ν, см–1
Рис. 3.1. ІЧ спектри ДМСО (1) і дейтерованого ДМСО (2)
При ізотопозаміщенні з метою віднесення частот коливання краще використовувати ізотопи, які мало відрізняються за масою. Невеликі зсуви частот у цьому випадку не спотворюють вигляд спектра, тому спектри двох ізотопних модифікацій можна легко порівняти. Так, наприклад, порівняння ІЧ спектрів ацетилацетонатів металів, які містили ізотопи 16О і 18О, дозволило провести віднесення смуг поглинання зв'язків СО і СС, які в цих комплексах мало відрізняються за формою та частотою (див. розд. 4). Ізотопний ефект широко застосовувався також для інтерпретації ІЧ спектрів комплексів металів із гідразидними лігандами (розд. 4).
Електронні ефекти (індукційний, мезомерний ефекти замісників, спряження) також значно впливають на частоту смуг поглинання. Так індукційний ефект замісників, кількісною мірою якого в аліфатичних системах є константа Тафта σ*, впливає на частоту зв'язків ν(С=О) і ν(Р=О) карбонільної та фосфорильної групи в сполуках R2C=O і R3P=O. Причина цього полягає в тому, що замісники, які мають позитивний індукційний ефект, наприклад, алкіли, знижують порядок зв'язку та зменшують частоту коливання ν(С=О). Замісники, які мають негативний індукційний ефект, наприклад, галогени, навпаки, збільшують частоту валентного коливання карбонільної та фосфорильної груп.
89
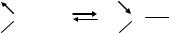
Сl |
СH3 |
|||
С |
|
О |
С+ |
О– |
|
||||
|
||||
R |
R |
|||
Частота коливання ν(С=О) у спектрі ацетону має значення 1715 см–1 (її розглядають як стандартну величину для зв'язку С=О), а в хлорангідридах кислот вона становить приблизно 1800 см–1. Заміна алкільного радикала на електронодонорні групи ОН, С6Н5 приводить до помітного зменшення частоти ν(С=О) унаслідок зміщення електронної густини зі зв'язку С=О на оксиген, що приводить до зменшення порядку зв'язку.
За наявності спряжених зв'язків відбувається перерозподіл електронної густини між кратними та простими (одинарними) зв'язками, що приводить до зниження частоти коливань кратних зв'язків і підвищення частоти коливань простих зв'язків. Так, наприклад, для спряжених зв'язків С=С низькочастотний зсув смуги поглинання дорівнює 20–40 см–1. Крім того, в ІЧ спектрах сполук, які мають спряжені зв'язки, зростає інтенсивність смуг валентних коливань кратних зв'язків і відбувається їх розщеплення.
Якщо спряження зв'язків С=С відбувається з С=О групою, то в ІЧ спектрах таких сполук спостерігається не лише зниження частот валентних коливань обох груп, але і значне зростання інтенсивності смуг поглинання С=С зв'язків. Фактично інтенсивність цих двох смуг стає приблизно однаковою. Характерним прикладом координаційних сполук, у яких спостерігається спряження зв'язків СС і СО, є β-дикетонати металів (див. наступний розділ). ІЧ спектри цих сполук показали, що в β-дикетонатному металоциклі відбувається вирівнювання зв'язків СО і СС, що пізніше було підтверджено методом рентгеноструктурного аналізу.
Утворення водневих зв'язків типу –OH…O–, –NH…N– чи >СО…HО– є характерним для багатьох сполук у конденсованому стані. Теплота утворення міжмолекулярного водневого зв'язку дорівнює 3–10 ккал/моль. Наявність водневих зв'язків приводить до утворення асоціатів різного складу: димерів, тримерів, полімерів тощо. При утворенні водневих зв'язків порядок зв'язку груп О–Н, N–H чи С=О знижується, наслідком чого є: зсув час-
90
тоти валентного коливання цих груп у низькочастотну область; збільшення інтенсивності смуг поглинання внаслідок підвищення полярності зв'язків; значне розширення смуг поглинання, які по суті складаються з кількох нерозділених смуг поглинання, що належать до асоціатів різного складу. Особливо широкі смуги поглинання спостерігаються для сполук, які містять у своєму складі групи ОН – води, спиртів і особливо карбонових кислот. Водневі зв'язки можуть бути внутрішньомолекулярними та міжмолекулярними.
Для системи О–Н…О енергія слабких і середньої сили водневих зв'язків може бути оцінена за рівнянням Н. Д. Соколова
Δν 3,8 10−3 ε,
ν0
де ν0 – частота (хвильове число) коливання ізольованого зв'язку ОН (см–1), ν – зсув частоти валентного коливання О–Н під впливом водневого зв'язку відносно ν0 в ІЧ спектрі зразка, ε – енергія водневих зв'язків (кДж).
Подібно до групи ОН, група N–H також здатна брати участь в утворенні як внутрішньо-, так і міжмолекулярних водневих зв'язків, але в силу меншої електронегативності атома нітрогену такі водневі зв'язки слабші, а величина низькочастотного зсуву та ширина смуг поглинання ν(NH), як правило, менші. Водневі зв'язки впливають також і на положення та форму смуги поглинання, що відповідає карбонільній групі. Частота коливань карбонільної групи, яка бере участь в утворенні водневих зв'язків знижується приблизно на 50 см–1, а сама смуга дещо розширюється. Форму смуги поглинання С=О часто використовують, щоб відрізнити її від інших коливань у тій же області спектра (наприклад С=N), які не розширені, оскільки не утворюють водневих зв'язків.
Щоб виключити вплив водневих зв'язків на ІЧ спектри, потрібно проводити дослідження речовин у неполярних апротонних розчинниках, які самі не здатні утворювати водневі зв'язки. Найчастіше з цією метою використовують CCl4, алкани чи хлороформ. При концентрації речовини порядку 10–2–10–3 моль/л концентрація асоціатів у розчині стає незначною. Так, в ІЧ спек-
91
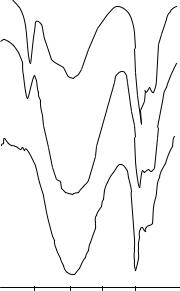
трах ізопропанолу широка смуга валентних коливань груп О–Н перебуває в діапазоні 3390–3320 см–1, вона свідчить про асоціацію молекул спирту. При розчиненні в ССl4 з'являється вузька смуга при 3630 см–1, типова для неасоційованих молекул ROН. Подальше зменшення концентрації приводить до зменшення інтенсивності смуги валентних коливань асоційованих ОН груп і збільшення інтенсивності вузької смуги неасоційованих молекул. Але така концентраційна залежність характерна тільки для ІЧ спектрів сполук з міжмолекулярними водневими зв'язками. Якщо водневі зв'язки мають внутрішньомолекулярний характер, як, наприклад, у о-саліциловому альдегіді, то частота та ширина смуги поглинання ОН груп не залежить від концентрації розчинів. Можливості дослідження координаційних сполук у розчинах обмежені їх поганою розчинністю органічних розчинниках, які використовуються для запису ІЧ спектрів.
3
2
1
3600 3400 3200 3000 ν, см–1
Рис. 3.2. ІЧ спектри ізопропанолу (1) і його розчинів у ССl4: 1:10 (2) і 1:50 (3)
92