

Госпиталка
.pdf
|
Болевой синдром. Повышение билирубина, АЛТ, АСТ. |
|
|
|
Лихорадка с ознобом. Тошнота, рвота. Слабость. Кожный зуд, желтуха. Утолщение |
|
ногтевых фаланг. Точечная чувствительность в районе желчного пузыря. |
Холангит |
Повышение билирубина, АЛТ, АСТ, амилазы. Нейтрофилия, лимфоцитоз. |
|
|
|
Расширение границ органа. Болевой синдром. Слабость, потеря веса. |
|
Тревожность, ухудшение внимания. Отеки нижних конечностей. Желтизна кожных |
Застойная |
покровов. Одышка и сопровождающий ее кашель. Воспаление лимфоузлов. |
печень |
Повышение билирубина, АЛТ, АСТ. |
|
|
|
Стремительное развитие. Слабость, потеря веса, изменение стула, |
|
диспепсические явления, боли в правом подреберье. Желтуха, сухость кожи и |
Рак печени |
слизистых, зуд. Температура субфебрильная. |
|
|
|
Диспепсия, болевой синдром, желтизна кожи и слизистых, лихорадка. |
|
Эритема,»сосудистые звездочки», тремор рук. Кал светлый, моча темная. Болевой |
|
синдром. Высокий билирубин, АЛТ, АСТ. Снижение альбумина и белка. |
Некроз печени |
Моноцитопения, эозинопения. |
|
|
|
Диспепсия. Желтушность кожи и склер. Снижение веса, увеличение живота. Боли. |
|
Эритема, «сосудистые звездочки», изменение формы пальцев и ногтей. Асцит, |
Цирроз |
энцефалопатия. Повышен билирубин. Снижен белок в крови. Нейтрофилия. |
|
|
Показаниями к трансплантации: терминальные фазы диффузных болезней печени, фульминантная печеночная недостаточность и новообразования печени.
Показанием к трансплантации печени является необратимая острая или терминальная стадия хронической печеночной недостаточности различной этиологии.

7. Болезнь Вильсона-Коновалова. Этиология, патогенез, клиника, диагностика, лечение
Болезнь Вильсона – Коновалова (синонимы гепатолентикулярная дегенерация, гепатоцеребральная дистрофия) – тяжелое прогрессирующее наследственное заболевание, передающееся по аутосомно-рецессивному типу, в основе которого лежит нарушение экскреции меди из организма, приводящее к избыточному накоплению этого микроэлемента в тканях и сочетанному поражению паренхиматозных органов (прежде всего печени) и головного мозга (подкорковых ядер)
Этиология: Причиной возникновения БВК являются мутации гена ATP7B, который локализован на 13 хромосоме в локусе 13q14.3 и кодирует медьтранспортирующую АТФ-азу Р-типа – ATP7B.
Патогенез:
Основными ферментами, обеспечивающими транспорт меди в организме, являются АТФ-аза 7А и АТФ-аза 7В.
мРНК АТФ-азы 7В обнаружена в гепатоцитах и капиллярах мозга. Этот фермент участвует в выведении меди организма (из крови в желчь) и из головного мозга в кровь. Именно его недостаток вызывает болезнь ВильсонаКоновалова.
Отсутствие АТФ-азы 7В нарушает выделение меди из головного мозга в кровь, из крови в желчь и далее с калом из организма. Ведущим звеном патогенеза является хроническая интоксикация медью. Медь накапливается в печени, селезенке, почках, головном мозге, роговице, хрусталике глаза и других органах. Накопление меди в печени приводит к некрозу гепатоцитов, воспалению, фиброзу, пролиферации желчных протоков и циррозу; в головном мозге – к некрозу нейронов с образованием полостей (кист).
Жалобы:
боли в животе различной локализации;
•изменение цвета кожи;
•носовые кровотечения;
•тремор и непроизвольные движения;
•слюнотечение, дизартрия, нарушение глотания;
•мигренеподобные головные боли;
•бессонница;
•депрессия;
•невротическоеповедение;
•изменения личности;
•психоз.
Лабораторные исследования [1-6]:

•общий анализ крови: лейкопения, нормохромная анемия, тромб ретикулоцитоз, ускоренная СОЭ.
•общий анализ мочи: при поражении почек можно микрогемаобнаружитьурию, незначительную протеинурию, гиперкальциурию.
•суточная экскреция мочи: гиперкупренилурия, признаки развившейся признаками: глюкозурией, аминоацидурией, фосфатурией, уратурией,
•биохимический анализ крови: снижение церулоплазмина и общей меди, уровней свободной меди (таблица 1), аминотрасфераз-50 раз); билирубин(в 1,5 повы более чем в 2 раза, преимущественно за счет прямой фракции; фосфатазы обычно повышен; может быть повышена гаммаглютамилтранспептидазы (ГГТП); гипоальбуминемия.
•коагулограмма: снижение протромбинового индекса, гипофибриногене тромбинового времени.
•пеницилламиновый тест: необходимо исследовать мочу, собранную приема 500 мг пеницилламина и через 12 часов. У пациентов- с Коновалова суточная экскреция меди будет повышаться до более 1 <50мкг/сут). У здоровых людей значительного увеличения экскре наблюдается.
УЗИ - гепатоспленомегалия+холестаз Эзофагогастродуоденоскопия: на наличие варикозно расширенных вен
желудка Электроэнцефалография – проводится пациентам с тяжелыми нарушен
стороны ЦНСэпилептическими приступами, регистрируетсяактивностьэпилептичес МРТ головного мозга: более информативно в диагностике, чем КТ г Характерны билатеральные очаги пониженной-15 ммплотностив диаметре3 в обла
базальных ганглиев (хвостатое ядро, скорлупа и бледный шар), зубчатых ядер и коры -мозжечкасимптом «морды гигантской панды»По мере[19]. прогрессирования процесса выявляются признаки диффузного атро головного мозга с равномерным расширением субарахноидальных желудочковой системы
Пункционная биопсия печени: 1. для морфологического исследования печени, где выявляются дистрофические изменения клеток, некрозы, инфильтрацию и фиброз различной степени выраженности; 2. Опреде меди в препарате печенипри ГЛД медь находится на000уровнемкг наг11сухого вещества печени.
Лечение:
полупостельныйрежим –при высокой активности и декомпенсации цирро диета, направленная на уменьшение поступленияограничиваютсямедиорганиз продукты с высоким содержанием меди (печень, креветки, орехи, шок
Современная патогенетическая терапия ГЛД основана на использова медьэлиминирующих препаратов [10,11], главным-пеницилламина,образом Дтриентина
солей цинка-пеницилламин.Д |
и триентин–хелатные комплексоны, образующие с |
прочные соединения, |
которые легко выводятся из органи |
Препаратом выбора при ГЛД -пеницилламинявляетсяД . Лечение начинают с неболь с постепенным увеличением ее до терапевтической, под контролем в Начальные дозы составляют-500 мг/сут250 с постепенным-7 (каждыедней)увеличение4 дозы на 250 мг до лечебной дозировки-1500 мг/сут,1000которая-4даетсяприемав 2

8. Тромбоцитопении у детей – эпидемиология, этиология, классификация, патогенез. Клиника, диагностика, дифференциальная диагностика геморрагических синдромов. Неотложная помощь при геморрагическом синдроме у детей и подростков. Основные принципы лечения тромбоцитопении у детей. Осложнения. Исходы. Профилактика. Особенности диспансерного наблюдения.
Клиника: геморрагический синдром с-пятнистымпетехиальнотипом кровоточивости в слизистые.
Носовые, маточные кровотечения + др локализации.
Идиопатическаятромбоцитопеническая пурпура(ИТП)– заб, в основе которого нарушение Тр звена вследствие-пении Тр
По течению: остр(до6 мес)/хроническое По периоду: обострения/клинической -ремиссии/клиниколабораторнойремиссии
Диагностика: геморрагический синдром, снижение тромбоцитов меньше 3 Кровотечение по Дьюку больше 4 минут(повышено)
Снижение или отсутствие ретракции кровянного сгустка
Диф. Диагностика с вторичной пурпурой (при гемобластозах, апласт гиперспленизме)

Лечение: режим постельный Стол 5
Лечение Тромбоцитопения при отсутствии геморрагического синдром При нарастании кожного геморрагического синдрома в процессе на и/или присоединении кровотечения показана иммуносупрессивная (преднизолон) как начальная терапия назначается в средней суточ соответствует 2 мг/кга в3 сутки)недели нпо 3 раза в день)с (6,учетом10, суточно14 биоритма—2/3 суточнойдозы ГК даются в утренние часы.
При достижении полной-лаборатклиникорной ремиссии доза преднизолона уме 5-10 мг в 3 дня до полной отмены. Снижение количества тромбоцито дозы ГК не является показанием к возврату прежней дозы. В слу клинической ремиссии продолжение терапии ГК в прежней дозе до но тромбоцитов нецелесообразно, так как пролонгированное лечение Г тромбоцитов и способствует развитию осложнений.
При неэффективности–цитостатики
В/в ИммуноглобулинG lkz ,kjrfls H”C ctktptyrb

экстренная |
спленэктомия (по витальным показаниям, прежде всего |
кровоизлиянии). |
|
Трансфузии |
тромбомассы не показанысенсибилизации и резкого пов |
образованияантитробоцитарных антител( однако в тяжелых. случаях можно, хватает на 1-2
сут)
Также в период тромбоцитопении с проявлениями геморрагическо ограничивается двигательный режим. Проводится симптоматическая те
ангиопротекторы—дицинон per os в/в;
ингибиторы фибринолиза—аминокапроновая кислота-0,5 г/кг0,2 в сутки per os, в/в
местные способы остановки кровотечений.
Диспансерное наблюдение при острой тромбоцитопенической пурпуре необходимо на протяжениилет.пятиПри хронической форме малыша наблюдают, будет по возрасту переведен во взрослую поликлинику.
Ребенку с тромбоцитопенической пурпуройаспирининеантикоагулянтыдают и/и антиагреганты, запрещено УВЧ, под запретом нитрофураны, УФО.
На протяжении-5 лет3 ребенку нельзя переезжать в страну/регион с др
Если геморрагический синдром минимально выражен, а количество 100 тыс / мкл, то запрещены внутримышечные инъекции детям.
Нужно планово осматривать ребенка, чтобы выявить хронически провести их санацию.
Необходима профилактикаОРВИ у детей.
Профилактические прививки больным тромбоцитопенической пурпуро только при ремиссии симптомов.
9. Гемофилии у детей – эпидемиология, этиология, классификация, патогенез. Клиника, диагностика, дифференциальная диагностика гемофилий у детей. Основные принципы лечения гемофилий. Неотложная помощь при геморрагическом синдроме у детей и подростков. Осложнения. Исходы. Медико-генетическая консультация. Особенности диспансерного наблюдения.
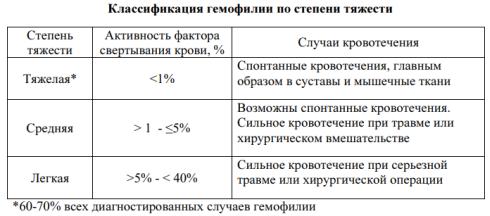
Гемофилия – это сцепленное с Х-хромосомой врожденное нарушение свёртываемости крови, вызванное недостаточностью или отсутствием фактора свертывания крови VIII (гемофилия А) или фактора IX (гемофилия В).
Распространенность гемофилии в большинстве стран составляет 10-14 больных на 100 000 мужчин. Гемофилия А (ГА) встречается чаще, чем гемофилия В (ГВ), и составляет 80-85%
Клинические проявления ГА и ГВ идентичны. Для клинической картины при гемофилии характерно развитие кровотечений

Как правило появляются после 6 мес.
Диагностика гемофилии. Данные семейного анамнеза примерно у 2/3 больных содержат указания на геморрагические проявления у близких родственников по женской линии (у мужчин, реже у женщин).
Физикальное обследование. При тяжёлой и среднетяжелой форме гемофилии выявляется кожный геморрагический синдром различной выраженности в виде множественных экхимозов и гематом. Возможны признаки поражения суставов в виде деформации, отека и локального повышения температуры кожи, позднее – признаки нарушения подвижности суставов, гипотрофия мышц конечности на стороне поражённого сустава, нарушение походки
+скрининг на наличие факторов перед операциями +пренатальная диагности и определение носителей
Лечение: Частота осмотра пациентов гематологом, ортопедом и стоматологом должна быть не менее 2 раз в год. Остальными специалистами – не менее 1 раза в год.
В основе лечения больных неосложненной формой гемофилии лежит специфическая заместительная терапия концентратами факторов свертывания крови VIII (при гемофилии А) или IX (при гемофилии В).
Препарат вводят-2 летс-31 2раза в неделю профилактической целью.
Инвалидность с детства, диспансеризация раз в год, б/х кров 2раза в год.

10.Геморрагический васкулит – эпидемиология, этиология, классификация, патогенез. Клиника, диагностика, дифференциальная диагностика геморрагического васкулита у детей. Основные принципы лечения геморрагического васкулита. Неотложная помощь при геморрагическом синдроме у детей. Осложнения, исходы, профилактика, особенности диспансерного наблюдения.
Болезнь Шенлейна–Геноха (БШГ) – васкулит, характеризующийся нетромбоцитопенической пурпурой с преимущественной локализацией на нижних конечностях и отложением IgA-депозитов в мелких сосудах (капилляры, венулы, артериолы). Типично вовлечение кожи, кишечника и клубочков почек; нередко сочетается с артралгиями или артритом.
Причины возникновения болезни остаются невыясненными. Болезнь Шенлейна– Геноха – это гиперергическая сосудистая реакция на различные факторы, чаще – инфекционные (стрептококк и другие бактерии, вирусы). В ряде случаев развитию болезни предшествуют вакцинация, воздействие пищевых и лекарственных аллергенов, укус насекомого, травма, охлаждение и т.д
В основе патогенеза БШГ лежит генерализованное иммунокомплексное повреждение сосудов микроциркуляторного русла с отложением гранулярных IgAдепозитов и последующей активацией системы комплемента, гемостаза. В результате ухудшаются реологические свойства крови, усиливается агрегация тромбоцитов и эритроцитов, развивается гиперкоагуляция. В сосудистой стенке возникают асептическое воспаление, деструкция, 7 тромбоз микрососудов, разрыв капилляров, что сопровождается геморрагическим синдромом
Рабочая классификация, принятая в РФ (А.А. Ильин, 1984), включает следующие параметры:
•фазы болезни (активная, стихания);
•клинические формы (простая, смешанная, смешанная с поражением почек);
•клинические синдромы (кожный, суставной, абдоминальный, почечный);
•степень тяжести (легкая, среднетяжелая, тяжелая);
• характер течения (острое, хроническое, рецидивирующее).
КЛИНИЧЕСКАЯ КАРТИНА
Начало острое. Температура тела субфебрильная, реже – фебрильная.
Поражение кожи Сыпь возникает у 100% больных, обычно – в начале болезни, реже – вслед за абдоминальным или суставным синдромом. Обильная (в тяжелых случаях
– сливная) сыпь локализуется преимущественно на коже нижних конечностей, ягодицах, вокруг крупных суставов, реже – на коже верхних конечностей, туловища, лица. По характеру сыпь мелкопятнистая или пятнисто-папулезная, геморрагическая, симметричная, как правило, сочетается с ангионевротическими отеками. Геморрагические элементы появляются волнообразно, оставляют после себя пигментацию, проходят бесследно.
Поражение суставов Наблюдается у 60–80% больных. Проявляется артралгиями, обратимым артритом преимущественно крупных суставов (коленных, голеностопных). Отмечаются болезненность, отек суставов и нарушение функции. Сохраняется от нескольких часов до нескольких дней.
Поражение ЖКТ Обусловлено отеком и геморрагиями в стенку кишки, брыжейку или брюшину. Боли в животе могут предшествовать кожному синдрому на 1-14 дней у 43% пациентов. Проявляется умеренными или сильными схваткообразными болями в животе. Болевые приступы могут повторяться многократно в течение дня и сопровождаться тошнотой, рвотой, жидким стулом с примесью крови. Могут развиться 8 аппендицит, холецистит, панкреатит, желудочно-кишечное кровотечение, язвы, инфаркты кишки, перфорация.
Поражение почек. В большинстве случаев протекает бессимптомно, что является основанием для проведения обследования в течение 6 месяцев после последнего эпизода кожного синдрома или других проявлений БШГ. Микрогематурия без протеинурии, как правило, протекает доброкачественно. Нарастающая протеинурия, развитие нефротического синдрома и/или почечной недостаточности отражают тяжелое течение заболевания. У 20% пациентов с нефротическим и нефритическим синдромом развивается терминальная почечная недостаточность; у 44-50% – артериальная гипертензия или хроническая почечная болезнь. Гистологически нефрит идентичен IgA-нефропатии и включает фокальный сегментарный пролиферативный гломерулонефрит и быстро прогрессирующий серповидный гломерулонефрит.
Течение БШГ чаще острое, с выздоровлением в течение 2 месяца от начала болезни, но может быть затяжным, рецидивирующим на протяжении 6 месяцев; редко длится в течение 1 года и более. Хроническое течение свойственно вариантам с нефритом Шенлейна–Геноха или с изолированным непрерывно рецидивирующим кожным геморрагическим синдромом.
Осложнения
•Инвагинация
•Кишечная непроходимость
•Перфорация кишечника с развитием перитонита
•При нефрите: острая почечная недостаточность или ХПН
ДИАГНОСТИКА Лабораторные исследования
Клинический анализ крови: умеренный лейкоцитоз с нейтрофилезом, эозинофилией, тромбоцитоз.
Биохимические и иммунологические исследования крови: повышение концентрации СРБ, IgА. 9