

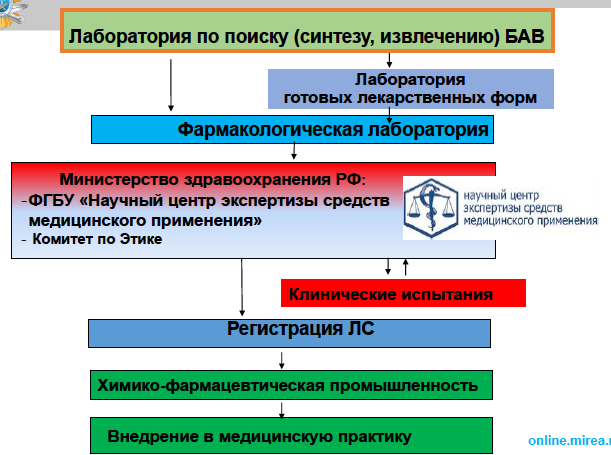
17. Охарактеризуйте этапы создания лекарственного препарата.
Поиск потенциального лекарственного средства
Тестирование лекарственного средства
Доклиническое исследование лекарственного средства
Подача заявления о государственной регистрации лекарственного препарата
Клинические испытания лекарственного средства
Экспертиза качества лекарственного препарата и отношения ожидаемой пользы к возможному риску
Включение лекарственного препарата в Государственный реестр лекарственных средств (РЛС)
Производство лекарственного препарата
Внедрение в медицинскую практику
R&D-период (research and development) начальный этап разработки ЛС
Создание препарата
Разработка препарата
Научное сопровождение препарата на этапе вывода на рынок
Что понимается под новым препаратом:
Новая молекула с новым механизмом действия
Новая молекула с известным механизмом действия
Модификация известной молекулы
Новая форма известного препарата
Разработка системы доставки лекарственного препарата
1. Поиск потенциального лекарственного средства:
- Теоретические модели (SAR – structure-activity relations)
- Экспериментальные модели – Скрининг (библиотеки веществ)
Разработка лекарственных средств:
Химический синтез препаратов
1. Получение препаратов из лекарственного сырья:
· животного происхождения
· растительного происхождения
· из минералов
2. Биотехнологическое получение - лекарственные вещества являются продуктами жизнедеятельности биообъектов:
использование микробиологического синтеза
методы генной инженерии
Замысел создания нового ЛВ. Возникает обычно в результате совместной работы ученых двух специальностей: фармакологов и химиков-синтетиков. Уже на этой стадии осуществляется предварительный отбор синтезированных соединений, которые, по мнению специалистов, могут быть потенциально биологически активными веществами.
Синтез предварительно отобранных структур. На этой стадии также осуществляется отбор, в результате которого вещества и т.д., не подвергаются дальнейшему исследованию.
Фармакологический скрининг и доклинические испытания. Основной этап, во время которого отсеиваются неперспективные вещества, синтезированные на предыдущем этапе.
Клиническая проверка. Ее выполняют только для перспективных БАВ, которые прошли все этапы фармакологического скрининга.
Регистрация препарата
Разработка технологии производства нового ЛВ и более рациональной ЛФ.
Подготовка нормативной документации, включающей способы контроля качества как самого ЛВ, так и его ЛФ.
Внедрение ЛВ в промышленное производство и отработка всех стадийного получения в заводских условиях.
18. Охарактеризуйте этапы испытания лекарственного средства.
Разработка и доклинические обычно 1-3 года
Этапы работ по результатам скрининга, как правило, закрыты. Исследователи проводят предварительный выбор вещества-лидера, а затем – окончательный выбор вещества-лидера. После этого вещество поступает в исследовательские группы, занимающиеся доклиническими испытаниями, подготовкой к клиническим испытаниям и созданием готовой лекарственной формы.
Тестирование лекарственного средства
Полученные вещества тестируются для подтверждения их фармакологической активности с применением биологических моделей различного организационного уровня: бесклеточный (молекулярный), клеточный, органы и ткани и на уровне целостного организма. Предварительное тестирование позволяет выявить перспективные молекулы для последующего их полного доклинического исследования на животных.
Доклинические испытания
Доклиническое исследование лекарственного средства -биологические, микробиологические, иммунологические, токсикологические, фармакологические, физические, химические и другие исследования лекарственного средства путем применения научных методов оценок в целях получения доказательств безопасности, качества и эффективности лекарственного средства (ФЗ от 12.04.2010 N 61-ФЗ "Об обращении лекарственных средств").
Нормативно правовые акты, регламентирующие проведение доклинических исследований лекарственных средств:
Федеральный закон от 12.04.2010 N 61-ФЗ «Об обращении лекарственных средств»;
Приказ Министерства здравоохранения и социального развития РФ от 23 августа 2010 г. N 708н "Об утверждении Правил лабораторной практики« (GLP)
Таким образом, для лекарственного вещества и разработанной для него лекарственной формы на этапе доклинических испытаний устанавливают специфическую активность в опытах на животных, изучают биологическую доступность, биологическую безвредность (острую и хроническую токсичность, местнораздражающее, аллергизирующее действие и т. д.), а также устойчивость при хранении. На основе полученных сведений составляется отчет и заключение о доклинических исследованиях будущего лекарственного средства. Затем клинические испытания
Федеральный закон от 12.04.2010 N 61-ФЗ "Об обращении лекарственных средств"
Статья 11. Доклиническое исследование лекарственного средства для медицинского применения
1. Доклиническое исследование лекарственного средства для медицинского применения проводится путем применения научных методов оценок в целях получения доказательств безопасности, качества и эффективности лекарственного средства.
2. Доклиническое исследование лекарственного средства для медицинского применения проводится в соответствии с правилами лабораторной практики, утвержденными уполномоченным федеральным органом исполнительной власти.
3. Для организации и проведения доклинического исследования лекарственного средства для медицинского применения разработчики лекарственных средств могут привлекать научно-исследовательские организации, образовательные учреждения высшего профессионального образования, имеющие необходимую материально-техническую базу и квалифицированных специалистов в соответствующей области исследования
4. Доклиническое исследование лекарственного средства для медицинского применения проводится по утвержденному разработчиком лекарственного средства плану с ведением протокола этого исследования и составлением отчета, в котором содержатся результаты этого исследования и заключение о возможности проведения клинического исследования лекарственного препарата для медицинского применения.
5. Проведение проверок соблюдения правил лабораторной практики и правовых норм использования животных при проведении доклинических исследований лекарственных средств для медицинского применения осуществляется уполномоченным федеральным органом исполнительной власти.
6. Результаты доклинического исследования лекарственного средства для медицинского применения могут быть представлены в уполномоченный федеральный орган исполнительной власти в установленном порядке в целях государственной регистрации лекарственного препарата.
Про клинические далее