

09_10_02
.pdfПовреждения 59
Почки |
Печень |
Почки обеспечивают активное выведение из орга низма с мочой ряда веществ с кислыми или основ ными свойствами, а также поддерживают концент рацию бикарбонатов крови. К главным механизмам уменьшения или устранения сдвигов КЩР крови, реализуемых нефронами почек, относят ацидоге нез, аммониогенез, секрецию фосфатов и K+,Na+ обменный механизм.
•Ацидогенез. Этот энергозависимый процесс, про текающий в эпителии дистальных отделов нефро на и собирательных трубочек, обеспечивает секре цию в просвет канальцев H + в обмен на реабсорбируемый Na+.
Количество секретируемого H+ эквивалентно его ко личеству, попадающему в кровь с нелетучими кис
лотами и H2CO3. Реабсорбированный из просвета канальцев в плазму крови Na+ участвует в регенера
ции плазменной гидрокарбонатной буферной сис темы.
•Аммониогенез, как и ацидогенез, реализует эпите лий канальцев нефрона и собирательных трубочек. Аммониогенез осуществляется путём окислительно го дезаминирования аминокислот, преимущественно (примерно 2/3) — глутаминовой, в меньшей мере — аланина, аспарагина, лейцина, гистидина. Образую щийся при этом аммиак диффундирует в просвет ка
нальцев. Там NH3+ присоединяет ион H+ с образова нием иона аммония (NH4+). Ионы NH4+ замещают Na+ в солях и выделяются преимущественно в виде
NH4Cl и (NH4)2SO4. В кровь при этом поступает эк вивалентное количество гидрокарбоната натрия, обес
печивающего регенерацию гидрокарбонатной буфер ной системы.
•Секреция фосфатов осуществляется эпителием дис тальных канальцев при участии фосфатной буфер ной системы:
Na2HPO4 + H2CO3 NaH2PO4 + NaHCO3
Образующийся гидрокарбонат натрия реабсорбиру ется в кровь и поддерживает гидрокарбонатный бу фер, а NaH2PO4 выводится из организма с мочой.
•К+,Na+ обменный механизм, реализуемый в дис тальных отделах нефрона и начальных участках со бирательных трубочек, обеспечивает обмен Na+ пер вичной мочи на K+, выводящийся в неё эпителиальными клетками. Реабсорбированный Na+ в жидких средах организма участвует в регене рации гидрокарбонатной буферной системы. K+,Na+ обмен контролируется альдостероном. Бо лее того, альдостерон регулирует (увеличивает) объём секреции и экскреции H+.
Таким образом, почечные механизмы устранения или уменьшения сдвигов КЩР осуществляются путём экскреции H+ и восстановления резерва гидрокарбонатной буферной системы в жидких средах организма.
Печень играет существенную роль в компенса ции сдвигов КЩР. В ней действуют, с одной сто роны, общие внутри и внеклеточные буферные системы (гидрокарбонатная, белковая и др.), с другой стороны, в гепатоцитах осуществляются различные реакции метаболизма, имеющие пря мое отношение к устранению расстройств КЩР.
•Синтез белков крови, входящих в белковую буфер ную систему. В печени образуются все альбумины, а также фибриноген, протромбин, проконвертин, про акцелерин, гепарин, ряд глобулинов и ферментов.
•Образование аммиака, способного нейтрализовать кислоты как в самих гепатоцитах, так и в плазме кро ви и в межклеточной жидкости.
•Синтез глюкозы из неуглеводных веществ — ами нокислот, глицерина, лактата, пирувата. Включе ние этих органических нелетучих кислот при обра зовании глюкозы обеспечивает снижение их содержания в клетках и биологических жидкостях. Так, молочная кислота, которую многие органы и ткани не способны метаболизировать, в гепатоци
тах примерно на 80% трансформируется в H2O и CO2, а оставшееся количество ресинтезируется в глюкозу. Таким образом, лактат превращается в
нейтральные продукты.
•Выведение из организма нелетучих кислот — глюку роновой и серной при детоксикации продуктов мета болизма и ксенобиотиков.
•Экскреция в кишечник кислых и основных веществ с жёлчью.
Желудок и кишечник
Желудок участвует в демпфировании сдвигов КЩР главным образом путём изменения секре ции соляной кислоты: при защелачивании жид ких сред организма этот процесс тормозится, а при закислении — усиливается. Кишечник спо собствует уменьшению или устранению сдвигов КЩР посредством:
•секреции кишечного сока, содержащего большое ко личество гидрокарбоната. При этом в плазму крови поступает H+;
•изменения количества всасываемой жидкости. Это способствует нормализации водного и электролит ного баланса в клетках, во внеклеточной и других биологических жидкостях и как следствие — норма лизации рН;
•реабсорбции компонентов буферных систем (Na+, K+, Ca2+, Cl–, HCO3–).
Поджелудочная железа способствует компенса ции сдвигов КЩР при помощи гидрокарбоната. Его секреция увеличивается при алкалозах и уменьшается в условиях ацидоза.

60 ПАТОЛОГИЯ Глава 2
КЛАССИФИКАЦИЯ РАССТРОЙСТВ КИСЛОТНО-ЩЕЛОЧНОГО РАВНОВЕСИЯ
Расстройства КЩР классифицируют по несколь ким критериям (табл. 2 7).
Ацидоз (МКБ: E87.2 Ацидоз) — форма наруше ния КЩР, характеризующаяся относительным или абсолютным избытком в организме кислот. В крови при ацидозе наблюдаются абсолютное или относительное повышение [Н+] и уменьше ние рН ниже нормы (условно — ниже средней величины рН, принимаемой за 7,39).
Алкалоз (МКБ: E87.3 Алкалоз) — форма нару шения КЩР, характеризующаяся относительным или абсолютным избытком в организме основа ний. В крови при алкалозе отмечается абсолют ное или относительное снижение [Н+] или уве личение рН (условно — выше средней величины рН, принимаемой за 7,39).
Газовый ацидоз характеризуется снижением рН кро ви и гиперкапнией (повышением рСО2 крови более 40 мм рт.ст.). При этом линейной зависимости между степенью гиперкапнии и клиническими признаками респираторного ацидоза нет. Последние во многом оп ределяются причиной гиперкапнии, особенностями ос новного заболевания и реактивностью организма па циента.
Газовый алкалоз характеризуется увеличением рН и ги покапнией (снижением рСО2 крови до 35 мм рт.ст. и более).
Негазовый ацидоз — наиболее частая и опасная фор ма нарушения КЩР. Такой ацидоз может наблюдать ся при сердечной недостаточности, многих типах ги поксии, нарушениях функций печени и почек по нейтрализации и экскреции кислых веществ, истоще нии буферных систем (например, в результате крово потери или гипопротеинемии), введении в организм веществ, содержащих избыток ионов водорода, и при других состояниях.
Таблица 2-7. Виды нарушений кислотно-щелочного равновесия
Негазовый алкалоз характеризуется повышением рН крови и увеличением концентрации бикарбоната в кро ви и в других жидкостях организма.
ВАРИАНТЫ СМЕРТИ КЛЕТОК*
Смерть клеток в организме может развиваться по двум принципиально различным путям: во первых, в результате необратимых (летальных) повреждений клетки под действием повреж дающих факторов; во вторых, без предваритель ного развития летальных повреждений клетки, а в ряде случаев и без каких либо поврежде ний, но в результате включения генетической программы смерти, предопределяющей гибель клетки.
НЕКРОЗ
Смерть клеток в результате действия повреж дающих факторов в ходе болезней, а также при летальных воздействиях экзогенных агентов, сопровождающихся активацией гидролитичес ких ферментов и приводящих к развитию нео братимых повреждений клеток, называется в патологии некрозом.
АПОПТОЗ
Смерть клеток без предварительных необратимых повреждений клеток, но в результате включе ния генетической программы, предопределя ющей их гибель, была названа запрограмми рованной клеточной смертью в 1964 г. [51]. Запрограммированная клеточная гибель встре чается в случаях, когда при развитии, росте и выживании тканей, а также при конструирова нии развивающихся структур в ходе морфоге
* Автор раздела «Варианты смерти клеток» — Е.А. Коган.
Критерии |
Виды нарушений КЩР |
|
|
Направленность изменений [H+] и |
Ацидозы, алкалозы |
рН |
|
Причины, вызвавшие нарушения |
Эндогенные, экзогенные |
КЩР |
|
Степень компенсированности |
Компенсированные, субкомпенсированные, некомпенсированные |
нарушений КЩР |
|
Причины и механизмы развития |
Газовые |
нарушений КЩР |
|
|
Негазовые метаболические, выделительные (почечные, желудочные, |
|
кишечные), экзогенные |
|
Смешанные (комбинированные) |
неза возникает необходимость избавиться от части клеток (в том числе от повреждённых и закончивших свой жизненный цикл). Возни кает парадоксальная ситуация: клетка жертву ет своей жизнью, идёт на самоубийство ради сохранения целого — ткани, органа, организ ма. Таким образом, биологическое значение программной клеточной гибели заключается в поддержании жизни.
Запрограммированная клеточная гибель явля ется составной частью таких биологических процессов, как эмбриональное развитие, мор фогенез и метаморфоз организмов. В много клеточных организмах баланс между запрог раммированной клеточной гибелью и митозом и обеспечивает тканевый гомеостаз.
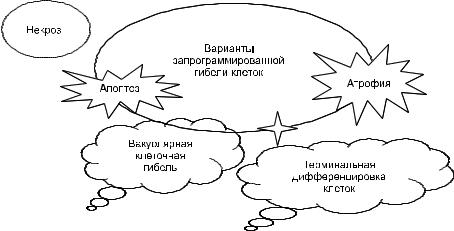
Термины «запрограммированная клеточная ги бель» и «апоптоз» не являются синонимами. Термин «запрограммированная клеточная ги бель» используют в литературе как в узком, так и в широком смысле этого слова.
•В узком смысле запрограммированная клеточ ная гибель противопоставляется апоптозу, поскольку первая совершается в норме в ходе развития организма и при обеспечении тка невого гомеостаза. В то же время апоптоз — самоубийство клетки — развивается в усло виях патологии.
•В широком смысле понятие «запрограммиро ванная клеточная гибель» подразумевает не только смерть клетки путём апоптоза, но и ряд других видов клеточной гибели: вакуолярная,
Повреждения 61
или аутофагическая, атрофическая смерть, а также гибель по достижении клеткой терми нальной дифференцировки (рис. 2 17).
Некроз
Некроз (от греч. necros — мёртвый) — омерт вение, гибель клеток и тканей в живом орга низме в ответ на повреждение, сопровождаю щееся активацией гидролитических ферментов и развитием аутолиза.
Понятие «некроз» является видовым по отно шению к более общему понятию «смерть». Не кроз — гибель части живого организма, тогда как целое — организм — остаётся живым. На против, термин «смерть» используется для обозначения прекращения жизнедеятельнос ти организма в целом. Территория некроза мо жет быть различной. Некроз может захватывать отдельные участки тела, целые органы, ткани, группы клеток и отдельную клетку [150]. В на стоящее время сформировалось также понятие о фокальном некрозе, когда речь идёт о гибели части клетки. Некроз развивается, как прави ло, при действии повреждающего фактора.
Некроз происходит только в условиях патоло гии, может иметь самостоятельное значение, а также завершать или входить в качестве одно го из важнейших элементов в такие патологи ческие процессы, как дистрофии, воспаление, расстройства кровообращения, опухолевый рост и др.
Ðèñ. 2-17. Виды клеточной гибели.

62 ПАТОЛОГИЯ Глава 2
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ НЕКРОЗА
Выделяют пять основных этиологических фак торов некроза: травматический, токсический, трофоневротический, аллергический и сосуди стый. Перечисленные факторы могут оказывать или непосредственное действие на ткань, или опосредованное — через сосудистую, нервную и иммунную системы. Этиологический прин цип и лёг в основу классификации видов некро за. По механизму действия этиологического фактора некроз может быть прямым и непря мым. Прямой некроз может быть травматичес ким и токсическим. Непрямой некроз — тро фоневротическим, аллергическим и сосудистым.
КЛАССИФИКАЦИЯ ВИДОВ НЕКРОЗА
Травматический некроз является результатом прямого действия на ткань физических (ме ханических, температурных, вибрационных, радиационных и др.) и химических (кислот, щелочей и др.) факторов.
Токсический некроз развивается при действии на ткани токсических факторов бактериаль ной и другой природы.
Трофоневротический некроз развивается при нарушении циркуляции и иннервации тка ней при заболеваниях центральной и пери ферической нервной системы. Примером трофоневротического некроза могут служить пролежни.
Аллергический некроз. Аллергический некроз является результатом иммунного цитолиза тканей в ходе реакций гиперчувствительнос ти немедленного и замедленного типов (ГНТ
иГЗТ соответственно). Классическим приме ром аллергического некроза при ГНТ с участием иммунных комплексов, содержащих комплемент, может служить фибриноидный некроз при феномене Артюса. Иммунный цитолиз с участием T лимфоцитов киллеров
имакрофагов приводит к развитию некроза ткани печени при хроническом активном ге патите.
Сосудистый некроз связан с абсолютной или относительной недостаточностью циркуля ции в артериях, венах и лимфатических со судах. Наиболее частая форма сосудистого некроза обусловлена нарушением кровообра щения в артериях в связи с их тромбозом, эм болией, длительным спазмом, а также с фун кциональным перенапряжением органа в условиях гипоксии. Недостаточная циркуля
ция в ткани вызывает её ишемию, гипоксию и развитие ишемического некроза, патогенез которого связан не только с гипоксическими, но и реперфузионными механизмами.
МЕХАНИЗМЫ НЕКРОЗА
Механизмы некроза (рис. 2 18) отличаются от механизмов аутолиза, они разнообразны, во многом зависят от характера повреждающего фактора и структурно функциональных осо бенностей клеток, тканей и органов, в которых развивается некроз. Конечный результат всех патогенетических механизмов некроза — воз никновение внутриклеточного хаоса. Из всего многообразия патогенетических путей некро за, вероятно, можно выделить пять наиболее значимых: 1) связывание клеточных белков с убиквитином, 2) дефицит АТФ, 3) генерация активных форм кислорода, 4) нарушение каль циевого гомеостаза и 5) потеря клеточными мембранами селективной проницаемости.
•Убиквитин — один из наиболее консерватив ных белков — в составе протеосом формиру ет ковалентные связи с остатками лизина по липептидных цепочек других белков. Синтез убиквитина, как и белков из семейства бел ков теплового шока, потенцируют различные повреждения. Так, в клетках ЦНС при болез ни Альцхаймера и болезни Паркинсона, а также в гепатоцитах при алкогольном пора жении печени обнаруживают протеосомы — комплексы белков с убиквитином. Такие
Ðèñ. 2-18. Механизмы некроза.
комплексы в гепатоцитах издавна известны как тельца Мэллори.
•Дефицит АТФ постоянно обнаруживается в гибнущих клетках. Долгое время полагали, что основной причиной некроза кардиомиоцитов при ишемии является снижение образования макроэргических соединений до определённо го уровня. В последние годы было показано, что в ишемическом повреждении участвуют и дру гие механизмы. Так, если ишемизированный миокард подвергнуть реперфузии, то некроти ческие изменения наступают гораздо быстрее и в больших масштабах. Описанные изменения были названы реперфузионными повреждени ями. Использование ингибиторов кальция (та ких как хлорпромазин) и антиоксидантов, не смотря на низкий уровень АТФ, уменьшает реперфузионные повреждения, что указывает на то, что для развития некроза одного дефи цита АТФ ещё недостаточно.
•Генерация активных форм кислорода (синглет ный кислород, супероксид анион радикал, анион гидроксила, пероксид водорода и др.) постоянно происходит в живых клетках. Всту пая во взаимодействия с липидами мембран, молекулами ДНК, вызывая оксидативный стресс, активные формы кислорода повышают проницаемость мембран, ингибируют катион ные насосы, потенцируют дефицит АТФ и из быток внутриклеточного кальция, что приво дит к развитию повреждения клетки и ткани. Наибольшее значение активные формы кисло рода играют в патогенезе некроза пневмоцитов при дистресс синдроме новорождённых, разви вающемся в результате оксигенотерапии, репер фузионных повреждений при инфаркте миокар да и некрозе гепатоцитов при передозировке парацетамола.
•Нарушения кальциевого гомеостаза характери зуются накоплением внутриклеточного каль ция в гибнущих клетках. В живых клетках внут риклеточная концентрация ионов кальция примерно в тысячу раз меньше по сравнению с внеклеточной. Инициальные изменения при повреждении связаны с нарушением работы катионных помп в связи с дефицитом АТФ. При этом кальций накапливается внутри кле ток, прежде всего в митохондриях. Происходит активация Ca2+ зависимых протеаз и фосфоли паз, что приводит к необратимым повреждени ям мембран (митохондриальных, цитоплазма тических), к ещё большим нарушениям их проницаемости и смерти клетки.
Повреждения 63
•Потеря способности к избирательной прони цаемости цитоплазматических мембран явля ется одним из характерных признаков некроза при воздействии комплемента, вирусных ин фекциях и гипоксических повреждениях. При этом происходит повреждение трансмембран ных белков, рецепторов и ферментных систем, регулирующих прохождение в клетку опреде лённых веществ. При воздействии комплемен та и перфоринов в цитоплазматическую мемб рану встраиваются протеиновые полимеразы. Литические вирусы также взаимодействуют с липидами мембран, встраивают в них белки вирусных капсидов, что приводит к разруше нию цитоплазматических мембран в момент выхода вируса из инфицированной клетки. В клетках, подвергшихся ишемии, нарушается расположение трансмембранных белков с фор мированием характерных белковых «гипокси ческих» уплотнений.
МОРФОГЕНЕЗ НЕКРОЗА
Некротический процесс проходит ряд морфо генетических стадий: паранекроз, некробиоз, смерть клетки, аутолиз.
Паранекроз — подобные некротическим, но обратимые изменения.
Некробиоз — необратимые дистрофические изменения, характеризующиеся преоблада нием катаболических реакций над анаболи ческими.
Смерть клетки, время которой установить трудно.
Аутолиз — разложение мёртвого субстрата под действием гидролитических ферментов по гибших клеток и клеток воспалительного инфильтрата.
КРИТЕРИИ СМЕРТИ КЛЕТКИ
Установление момента смерти клетки, необра тимого её повреждения имеет важное теорети ческое и клиническое значение при решении вопроса о жизнеспособности тканей, подлежа щих хирургическому удалению, а также в транс плантологии. Однозначного ответа на этот вопрос пока не существует. В токсикологической практике критерием жиз неспособности тканей является, например, со хранность способности клеток к пролифера ции. Но можно ли считать клетку погибшей, если она находится в фазе покоя G1, может диф ференцироваться и оставаться жизнеспособной ещё длительное время, как это и происходит с
64 ПАТОЛОГИЯ Глава 2
большинством клеток многоклеточных орга низмов. Предлагается также использовать ме тод in vitro для установления гибели клеток и тканей, основанный на захвате ими различных красителей (трипанового синего и др.). Метод захвата краски также не может служить досто верным критерием оценки смерти клетки, так как скорее связан с повреждением цитоплаз матической мембраны, а не с некрозом. Как видно, достоверных функциональных тестов для установления момента смерти клеток пока ещё не разработано.
Для определения смерти клетки наиболее час то используют морфологические критерии. Та кими достоверными критериями необратимо сти повреждения клетки в ЭМ являются отложения в митохондриях электроноплотных депозитов, содержащих белки и соли кальция, и разрушение их внутренних мембран. В СМ изменения в структуре клетки становятся ви димыми лишь на стадии аутолиза. Поэтому, говоря о микроскопических признаках некро за, мы фактически говорим о морфологичес ких изменениях на стадии аутолиза.
МОРФОЛОГИЯ НЕКРОЗА
МАКРОСКОПИЧЕСКИЕ ПРИЗНАКИ НЕКРОЗА
Общими для всех форм некроза являются из менения цвета, консистенции и в ряде случа ев — запаха некротических тканей. Некроти зированная ткань может иметь плотную и сухую консистенцию, что наблюдается при коагуляционном некрозе. Ткань при этом мо жет подвергнуться мумификации. В других случаях мёртвая ткань дряблая, содержит боль шое количество жидкости, подвергается мио маляции (от греч. malakas — мягкий). Такой некроз по консистенции называется коллик вационным.
Цвет некротических масс зависит от наличия примесей крови и различных пигментов, а так же обусловлен развитием на границе между мёртвой и живой тканью зоны демаркацион ного воспаления, имеющей красно бурый цвет. Мёртвая ткань бывает белой или желтоватой, нередко окружена красно бурым венчиком. При пропитывании некротических масс кровью они могут приобретать окраску от красной до бурой, жёлтой и зелёной (в зависимости от преобладания в них тех или иных гемоглобин ных пигментов). В некоторых случаях фокусы некроза прокрашиваются жёлчью. При гнило
стном расплавлении мёртвая ткань издаёт ха рактерный дурной запах.
По цвету инфаркт может быть белым (селезён ка, головной мозг), белым с геморрагическим венчиком (сердце, почки) и красным (гемор рагическим). Геморрагический венчик форми руется за счёт зоны демаркационного воспале ния, которая закономерно возникает на границе мёртвых и живых тканей. Красный цвет инфаркта обусловлен пропитыванием не кротических тканей кровью, как это бывает при инфарктах лёгкого на фоне хронического ве нозного полнокровия.
МИКРОСКОПИЧЕСКИЕ ПРИЗНАКИ НЕКРОЗА
Микроскопические признаки некроза (рис. 2 19, 2 20, 2 21, 2 22 на вклейке) выявляют ся как в изменениях ядра, так и цитоплазмы. Ядра последовательно подвергаются сморщи ванию (кариопикноз), распаду на глыбки (ка риорексис) и лизируются (кариолизис). Эти изменения ядер связаны с активацией гидро лаз — рибонуклеаз и дезоксирибонуклеаз. В цитоплазме происходит денатурация и коагу ляция белков, обычно сменяемая колликваци ей. Коагуляция цитоплазмы сменяется распа дом её на глыбки (плазморексис) и лизисом органелл (плазмолизис). При фокальных из менениях говорят о фокальном коагуляцион ном некрозе и фокальном колликвационном некрозе (баллонная дистрофия).
Некроз развивается не только в паренхиматоз ных элементах тканей и органов, но и в их стро ме. При этом происходит разрушение как кле ток, так и нервных окончаний и компонентов межклеточного матрикса. Расщепление рети кулярных, коллагеновых и эластических воло кон происходит с участием нейтральных про теаз (коллагеназ, эластазы), гликопротеинов — протеаз, липидов — липаз. При СМ обнару живаются распад, фрагментация и лизис рети кулярных, коллагеновых и эластических воло кон (эластолизис), в некротизированной ткани нередко откладывается фибрин. Описанные изменения характерны для фибриноидного некроза. В жировой ткани некроз носит спе цифические черты в связи с накоплением в некротических массах жирных кислот и мыл, что ведёт к образованию липогранулём.
Развитие некроза, как правило, сопровожда ется возникновением местного демаркаци онного острого воспаления, возникновение которого связывают с выделением некротизи
рованной тканью провоспалительных субстан ций. Природа этих веществ пока недостаточно изучена. Имеются указания на генерацию по гибающими клетками лейкотриенов — мощных медиаторов воспаления, образующихся при перекисном окислении липидов. Кроме того, компоненты, освобождающиеся при поврежде нии митохондрий, являются сильными акти ваторами системы комплемента. Сама воспа лительная реакция на некроз может вызвать дополнительные повреждения сохранившихся в зоне демаркационного воспаления клеток и тканей. Это особенно важно при инфаркте миокарда, когда некроз кардиомиоцитов обна руживается не только в зоне некроза, но и в зоне перифокального воспаления, что значи тельно увеличивает площадь некроза миокар да. Повреждение кардиомиоцитов в зоне де маркационного воспаления обусловлено как реперфузией, так и действием клеток воспали тельного инфильтрата — прежде всего ПЯЛ и макрофагов, генерирующих протеазы и актив ные формы кислорода.
Реакция на некроз может быть не только мест ной, но и системной. Системная реакция на некроз связана с синтезом клетками печени двух белков острой фазы воспаления — С ре активного белка и плазменного белка SAA. Концентрация С реактивного белка повышает ся в плазме при различных видах повреждения. Этот белок аккумулируется в некротических массах и может активировать комплемент по классическому пути и инициировать разви тие демаркационного воспаления. Роль бел ка SAA, который может попадать в кровь из очагов некроза, связана с опсонизированием хроматина. Этот белок может стать белком предшественником при формировании АА амилоида.
УЛЬТРАСТРУКТУРНЫЕ ПРИЗНАКИ НЕКРОЗА
ЭМ признаки некроза отражают изменения органелл клетки.
•Ядро — агрегация хроматина, фрагментация фибрилл, полное разрушение.
•Митохондрии — набухание, уменьшение плотности гранул матрикса, образование в нём агрегатов неправильной формы, отложе ние солей кальция.
•Эндоплазматическая сеть — набухание, фраг ментация и распад мембранных структур.
•Полисомы и рибосомы — распад полисом, от деление рибосом от поверхности цистерн,
Повреждения 65
уменьшение чёткости контуров и размеров,
а также количества рибосом.
•Лизосомы — агрегация мелких плотных гра нул матрикса и его просветление, разрыв мембран.
•Цитоплазматический матрикс — исчезнове ние гранул гликогена, снижение активности ферментов [134].
ИСХОДЫ НЕКРОЗА
Нередко некроз ткани или органа имеет небла гоприятный исход и приводит больного к смер ти. Таковы, например, инфаркты миокарда, го ловного мозга, некроз коркового вещества почек, некроз надпочечников, прогрессирую щий некроз печени, панкреонекроз. К неблагоприятным исходам некроза относится также гнойное расплавление, что может быть причиной прогрессирования гнойного воспа ления вплоть до генерализации инфекционно го процесса и развития сепсиса.
Благоприятные исходы некроза связаны с про цессами отграничения и репарации, начина ющимися и распространяющимися из зоны де маркационного воспаления. К ним относятся: организация или рубцевание (замещение не кротических масс соединительной тканью), инкапсуляция (отграничение некротизирован ного участка соединительнотканной капсулой; при этом некротические массы петрифициру ются [пропитываются солями кальция] и ос сифицируются [образуется кость]). На месте колликвационного некроза образуется мезо глиальный рубчик (при небольших размерах некроза) или киста.
КЛИНИКО-МОРФОЛОГИЧЕСКИЕ ФОРМЫ НЕКРОЗА
Клинико морфологические формы некроза выделяют в зависимости от особенностей мор фологических и клинических проявлений той или иной формы некроза, что определяется его этиологией и структурно функциональными особенностями органа, в котором некроз раз вивается. Различают следующие виды некро за: коагуляционный, казеозный, колликваци онный, жировой, инфаркт, гангрену, секвестр и пролежень.
Коагуляционный некроз развивается при низ кой активности гидролитических процессов, высоком содержании белков и низком содер жании жидкости в тканях. Примером могут
66 ПАТОЛОГИЯ Глава 2
служить восковидный, или ценкеровский, не кроз мышц при брюшном и сыпном тифе; тво рожистый некроз при туберкулёзе, сифилисе, проказе и лимфогранулематозе; фибриноид ный некроз при аллергических и аутоиммун ных заболеваниях.
Казеозный (творожистый) некроз является разновид ностью коагуляционного некроза и получил своё на звание за сходство по консистенции, цвету и виду с творогом. При химическом анализе в некротических тканях (помимо большого количества преципитиро ванного белка) обнаруживаются и липиды. Встреча ется при туберкулёзе. Некрозу подвергаются скопле ния клеток моноцитарного происхождения — гранулёмы. Патогенез казеозного некроза объясня ется действием на клетки гранулёмы фактора, выде ляющегося самими клетками гранулёмы — ФНОα , а также факторов микобактерии.
Колликвационный некроз развивается в тканях, богатых жидкостью с высокой активностью гидролитических ферментов. Классическим примером может служить очаг серого размяг чения головного мозга. Для очагов реперфу зии в демаркационной зоне инфаркта миокар да также характерно развитие колликвационного некроза, которому может предшествовать коагуляционный некроз кар диомиоцитов.
Гангрена (от греч. gangrania — пожар) — некроз тканей, соприкасающихся с внешней средой. Ткани имеют чёрную окраску в результате об разования сульфида железа из железа Hb и се роводорода воздуха. Гангрена может развивать ся в различных частях тела, лёгких, кишечнике, матке. Имеется три разновидности гангрены — сухая, влажная и пролежень.
Сухая гангрена. Ткани при сухой гангрене мумифици руются, на границе с сохранной живой тканью чётко определяется зона демаркационного воспаления. Встречается в конечностях и на теле при атероскле розе, отморожениях и ожогах, болезни Рейно и виб рационной болезни, при тяжёлых инфекциях.
Влажная гангрена возникает в тканях при действии гнилостных микроорганизмов. Ткань набухает, ста новится отёчной, издаёт зловонный запах, демарка ционная зона не определяется. Влажная гангрена встречается в лёгких, кишечнике и матке. У ослаб ленных корью детей влажная гангрена может раз виться на коже щёк, промежности и называется но мой (от греч. nome — водяной рак).
Пролежень является разновидностью гангрены тро фоневротического генеза. Возникает в местах наи большего давления у ослабленных больных, страда ющих сердечно сосудистыми, инфекционными, онкологическими и нервными заболеваниями. Ло кализуются пролежни обычно на участках тела, под вергающихся у лежачих больных наибольшему дав лению.
Секвестр — участок мёртвой ткани, который не подвергается аутолизу, не замещается соедини тельной тканью и свободно располагается сре ди живых тканей. Секвестры обычно вызывают развитие гнойного воспаления и могут удалять ся через образующиеся при этом свищевые ходы. Секвестрации чаще подвергается костная ткань, однако секвестры, хотя и редко, но мо гут обнаруживаться и в мягких тканях.
Инфаркт (от лат. infarcire — начинать, набивать)
— сосудистый некроз (ишемический). Причи ны инфаркта — тромбоз, эмболия, длительный спазм артерий и функциональное перенапря жение органа в условиях гипоксии (недостаточ ности коллатерального кровообращения). Раз личают инфаркты по форме и цвету. Форма инфаркта зависит от ангиоархитектоники орга на и развитости коллатерального кровообраще ния и может быть клиновидной и неправиль ной. Клиновидная форма инфаркта характерна для органов с магистральным типом ветвления сосудов и со слабо развитыми коллатералями (селезёнка, почка, лёгкое). Неправильная фор ма инфаркта наблюдается в органах с рассып ным или смешанным типом ветвления артерий (миокард, головной мозг).
Апоптоз
Термин апоптоз (от греч. apoptosis — листо пад) предложен в 1972 г. [46] для обозначения особого вида запрограммированной смерти от дельных клеток, морфологически отличного от некроза, который авторы образно сравнили с падением увядших листьев с деревьев.
ЭТИОЛОГИЯ АПОПТОЗА
Причины и условия развития апоптоза много образны. Апоптоз закономерно развивается как в норме, так и при патологии в ходе под держания тканевого гомеостаза, развития им мунных реакций, элиминации повреждённых клеток, при старении.
АПОПТОЗ В НОРМЕ
Основная биологическая роль апоптоза в нор ме — установление равновесия между процес сами пролиферации и гибели клеток (рис. 2 23), что в одних ситуациях обеспечивает ста бильное состояние организма при сбалансиро ванности пролиферации и апоптоза клеток, в других — рост при преобладании пролифера

Ðèñ. 2-23. Апоптоз и пролиферация.
ции над апоптозом, в третьих — атрофию тка ней (вследствие относительного усиления апоп тоза по сравнению с пролиферацией).
В норме апоптоз наблюдается на стадиях пре имплантации и имплантации плодного яйца, органогенеза, а также морфогенеза. Исчезно вение клеток путём апоптоза хорошо докумен тировано при инволюции мюллерова и воль фова протоков, межпальцевых перепонок, при формировании просветов в полостных органах (например, в сердце). Апоптоз наблюдается
Повреждения 67
при атрофии зрелых тканей под влиянием или при отмене эндокринных стимулов при росте и старении организма. В качестве примеров апоптоза в норме могут быть приведены возра стная атрофия тимуса, возрастная инволюция ткани эндометрия и предстательной железы, МЖ после прекращения лактации. Классичес ким примером может служить апоптоз В и T лимфоцитов после прекращения действия на них стимулирующего действия соответствую щих цитокинов при завершении иммунных ре акций.
АПОПТОЗ В ПАТОЛОГИИ
Значение апоптоза в патологии велико. Пола гают, что все этиологические факторы, вызы вающие некроз, в небольших дозах и при крат ковременном воздействии способны вызывать и апоптоз (термические факторы, радиация, цитотоксические противораковые ЛС, гипок сия).
ПАТОГЕНЕЗ АПОПТОЗА
Патогенез апоптоза (рис. 2 24) включает био химические и генетические механизмы его ре гуляции.
Ðèñ. 2-24. Механизмы апоптоза. ФНОα — фактор некроза опухоли α ; Fas — поверхностный рецептор семейства ФНОα ;
Fas-L — лиганд Fas; ФНОα -Р1 — рецептор для ФНОα ; FADD — домен смерти, ассоциированный с Fas; TRADD — домен смерти, ассоциированный с ФНО; RIP — взаимодействующий с рецептором белок; RAIDD — ассоциированный с RIP белок с доменом смерти; FLIP — белок-ингибитор каспазы; TRAF-2 — ассоциированный с ФНО белок 2, активирует фактор транскрипции NF-kB; p53 — антионкоген p53 [ïî 7].
68 ПАТОЛОГИЯ Глава 2
БИОХИМИЧЕСКИЕ МЕХАНИЗМЫ РЕГУЛЯЦИИ
АПОПТОЗА
Биохимические механизмы регуляции апопто за условно могут быть разделены на четыре группы.
•Расщепление белков цистеиновыми протеа зами — каспазами.
•Сшивание белков трансглутаминазами в единую связанную сеть и обезвоживание кле ток за счёт действия селективных транспорт ных систем, регулирующих обмен ионов ка лия, натрия, хлора и воды. Высказываются мнения об участии в процессах конденсации
цитоплазмы белков цитоскелета, прежде всего β тубулина, усиление синтеза которого отме чается в клетках при апоптозе.
•Разрушение ядра кальций/магнийзависимой эндонуклеазой, расщепляющей молекулы ДНК в участках между нуклеосомами, что приводит к формированию однотипных по размерам фрагментов ДНК. Масса этих фраг ментов кратна массе одной нуклеосомы, со стоящей из 200 пар оснований, а каждый фрагмент содержит от одной до нескольких нуклеосом. Своеобразное расщепление ДНК при апоптозе имеет и своё морфологическое выражение в виде особой структуры хрома тина.
•Повреждение структуры клеточной мембра ны, сопровождающееся перемещением фос фатидилсерина с внутренней на наружную поверхность цитолеммы.
Изменение химической организации цитолем мы при апоптозе имеет решающее значение в распознавании и немедленном фагоцитозе апоптозных телец соседними клетками, что предупреждает попадание различных биологи чески активных субстанций в окружающую сре ду и тем самым предотвращает воспалительную и другие патологические реакции.
ГЕНЕТИЧЕСКИЕ МЕХАНИЗМЫ РЕГУЛЯЦИИ АПОПТОЗА
Большая группа генов и их белковых продук тов регулирует процессы апоптоза в клетках (см. рис. 2 24). В соответствии с фазами (ста диями) апоптоза эти гены и продукты их эксп рессии условно можно подразделить на 4 груп пы: 1) передающие сигнал от клеточной мембраны в клетку, 2) передающие сигнал внут ри клетки — контролирующие и интегрирую щие, 3) осуществляющие апоптоз, 4) регули рующие фагоцитоз апоптозных телец.
1. Индуцирующие апоптоз сигналы, как пра вило, поступают в клетку через клеточные мембраны. Ряд веществ посредством взаимо действия с соответствующими рецепторами клеточной мембраны может передавать нега тивный сигнал, блокирующий апоптоз (не которые гормоны, факторы роста и др.), дру гие — позитивный, индуцирующий апоптоз (Fas лиганды, ФНОα , ТФРβ и др.). Отметим, что отмена сигналов, блокирующих апоптоз, способна его индуцировать. Некоторые про апоптогенные факторы воздействуют сразу на внутриклеточные структуры (например, глю кокортикоиды вызывают апоптоз путём вза имодействия с соответствующими ядерными рецепторами).
2. Внутриклеточная фаза передачи апоптоген ного сигнала. На этой стадии наибольшее значение имеет семейство генов bcl, а также bcl2 связывающий белок Apaf1, каспаза 9, онкобелки p53, Rb, c vec, c fos, c jun.
•Члены семейства bcl2, расположенные в митохонд риях, регулируют проницаемость наружной мито хондриальной мембраны и тем самым могут способ ствовать или препятствовать выходу из митохондрий цитохрома С — важнейшего триггера апоптоза.
•Члены семейства, индуцирующие апоптоз, носят на звание промоторов апоптоза, к ним относятся bах, bаd и др.
•Члены семейства bcl2, препятствующие апоптозу, на зываются ингибиторами апоптоза и представлены bcl2, bcl XL и др.
•Все члены данного семейства способны взаимодей ствовать друг с другом и образовывать гетеродимеры, при этом наличие в составе гетеродимеров промото ров апоптоза является решающим в плане их функ циональной активности. Гетеродимеры — промото ры апоптоза из семейства bcl2 вызывают повышение проницаемости наружной мембраны митохондрий и высвобождение цитохрома С, что считается одним из наиболее ранних событий апоптоза.
3. Протеолиз. Высвободившийся из митохон дрий цитохром С взаимодействует с активи рующим протеазы проапоптозным фактором Apaf1 и тем самым индуцирует начало проте олитического каскада в клетке, определяю щего её смерть. Протеолитические фермен ты, участвующие в работе протеолитического каскада, могут быть подразделены на конт ролирующие и интегрирующие и представ лены семейством цистеиновых протеаз — кас паз. Каспазы расщепляют белки цитоскелета и матрикса клетки, но основной их мишенью являются ядро и ядерные белки, участвующие в транскрипции, репликации и репарации ДНК. В частности, каспаза 3 превращает не