

6 курс / Эндокринология / Stroev_Endokrinologia_podrostkov
.pdfния — гипоталамическим синдромом пубертатного периода {болезнью Симпсона-Пейджа), при ко-
тором также наблюдается ожирение со стриями. Но при болезни Симпсона-Пейдэюа ожирение равно-
мерное, рост высокий, половое развитие нормальное, нет остеопороза и признаков объемных процессов, стрии розовые, без атрофии. Болезнь СимпсонаПейджа часто спонтанно регрессирует, уровни АКТГ
икортизола при нем повышены умеренно.
Убомжей-подростков, злоупотребляющих алкоголем, может развиваться кушингоидная внешность с лунообразным синюшным лицом, ожирением андроидного типа, мышечной слабостью и даже со стриями в области живота (псевдосиндром Иценко-Кушинга).
Уних может обнаруживатьсяумеренный гиперкортизолизмснарушениемциркадногоритма секреции, однако лишениеихалкоголя быстроустраняет кушингоидную симптоматику(Балаболкин М.И.,1998).
Удевушек при поликистозе яичников (синдром Штейна-Левенталя) нет атрофии мышц и остеопороза, месячныесохранены,17-ОКС нормальные.
При скрываемой беременности нет месячных, уро-
вень АКТГ слегка повышен, гипофиз в объеме может увеличиваться, ноявногоизбытка кортизола нет, а специальные тесты на беременность и УЗИ матки устанавливают правильный диагноз.
Передозировка глюкокортикоидов легко распоз-
нается по анамнезу.
Специальные исследования позволяют исключить
АКТГ-продуцирующие опухоли (тимома, рак бронха).
Изредка при современных интраскопических методахисследованиябрюшной полости случайно(1,4-8,7% больных) выявляются округлой формы опухоли надпочечников — инциденталомы — без явных проявлений гормональной активности (иногда—с признаками
субклинического синдрома Иценко-Кушинга), но в
2-3 % случаев склонных к озлокачествлению (Воро- хобина Н.В., Силышцкий П.А. с соавт., 2003; Трофимова Т.Н. с соавт., 2000).
Исходы заболевания и прогноз. У подростков ис-
тинный синдром Иценко-Кушинга развивается постепенно, течет очень тяжело и без лечения всегда заканчивается летально.
Прогноз усугубляется склонностью к инфекциям, к пирамидным и стволовым синдромам, к атаксии, нистагму, гидроцефалии, эпилепсии.
Различные осложнения может дать артериальная пшертензия.
При легкой формесиндрома прогноз более оптими- стичен—послерадикальноголечениявозможновыз-
доровление, нопри средней тяжести и тяжелой формах изменения внутренних органов необратимы.
Особенно неблагоприятен прогноз при синдроме эктопической гиперпродукции АКТГ, вызванном ра-
ком любой локализации, а также при гиперкортицизме из-за рака надпочечника.
После оперативного лечения может развиться пангипопитуитаризм, синдром Нельсона или хроническая недостаточность коры надпочечников.
Лечение. Диета должна быть субкалорийной (не более 1800 ккал) с ограничением поваренной соли и легкоусвояемых углеводов.
Длительная лекарственная терапия синдрома Иценко-Кушинга направлена на подавление синтеза
АКТГ и кортизола парлоделом (бромокриптином), бромэргоном, абергином, достинексом; ГАМК-ер- гическими препаратами — дифенином , конвулексом, аминалоном, фенибутом; антисеротониновыми средствами — перитолом, ципрогептадином, метерголином, каберголином, особенно после луче-
вого лечения. В последние годы с успехом используют ритансерин, который является выраженным антагонистом серотониновых рецепторов 2-го типа и может нормализовать секрецию АКТГ и кортизола.
Используют также разрушители клеток надпочечни-
ков — митотан, хлодитан, лизодрен, которые вызывают «медикаметозную адреналэктомию», а также блокаторы стероидогенеза, блокирующие превращение холестерина в прегненолон (аминоглутетемид, элиптен, мамомит, ориметен). Угнетают синтез кортизола также трилостан, производные имидазола (кетоконазол, этомидат). Эффективноснижает продукцию кортизола метирапон — ингибитор 11в-гидроксила- зы.
Многие эти препараты токсичны, поэтому парал-
лельно дают эссенциале-форте, карсил, витамины
(кроме аскорбиновой кислоты, которая стимулирует синтез кортикостероидов!).
По показаниям назначают верошпирон, альдактон, триамтерен, салуретики, антагонисты кальция, анаболики (ретаболил, метандростенолон, неробол).
При остеопорозе показано ношение корсета, препараты кальция, кальцитрин (тирокальцитонин), препараты витамина D3 (рокальтрол, вигантол, остеохин, оксидевит).
При стероидном диабете показана диета № 9 по
М. Певзнеру с пероральными сахароснижающими средствами (инсулин требуется редко).
Иммунодефицит исправляется тималином, Т-ак- тивином и другими препаратами.
127

Радикальное лечение — лучевое и хирургическое. При легких и среднетяжелых формах болезни Ицен-
ко-Кушинга более показана гамматерапия гипофиза
с эффектом через 1-2 года. У подростков лучшим методом лучевой терапии считаются протонотера-
пия гипофиза в сочетании с односторонней адрена-
лэктомией. При этом особое значение имеет профилактика послеоперационного гипокортицизма, так как второй, интактный, надпочечник обычно бывает атро-
фированным. При болезни Иценко-Кушинга иногда применяют имплантацию в гипофиз радиоигл (итт-
риевых, золотых).
При операциях на гипофизе используют транссфеноидальный доступ. Методом выбора при болезни Иценко-Кушинга является селективная транссфеноидальная аденомэктомия.
При тяжелом синдроме удаляют оба надпочечника, затем проводят гамматерапию гипофиза во из-
бежание развития его реактивной опухоли (синдром Нельсона) с картиной надпочечниковой недостаточности, глазными и нервными нарушениями.
После двусторонней адреналэктомии требуется пожизненная заместительная терапия глюкокортикоидами (преднизон, преднизолон, кортизонацетат, кортизон-гемисукцинат).
Профилактика. Необходимо беречься от инфекций, простуд, травм головы, избегать воздействия на организм мутагенов.
Следует остерегаться ранней половой жизни и ранних беременностей (особенно у несовершеннолетних), бесконтрольного использования гормонов, анаболиков, допингов, контрацептивов.
Лечение различных болезней курсами глюкокортикоидов должно производиться с принятием необходимых мер против развития ятрогенного гиперкортицизма и синдрома отмены.
Диспансеризация. Диспансерная группа — Д-3. Больные с гиперкортицизмом нуждаются в пожизненном наблюдении эндокринологом, невропатологом, окулистом, кардиологом, а попоказаниям—и ортопедом. С диспансерного учета больные подростки не сни-
маются.
После двусторонней адреналэктомии при кортикостероме и при развитии тотальной гипофизарной недостаточности — пангипопитуитаризма — вследствие оперативного вмешательства на гипофизе при болез-
ни Иценко-Кушинга показана пожизненная заместительная глюкокортикоидная терапия (см. Хро-
ническая недостаточность коры надпочечников).
Вопросы экспертизы. Группа здоровья — 5. Все подростки с синдромом Иценко-Кушинга подлежат
128
направлению на МСЭК для определения группы инвалидности.
При редком полном регрессе могут быть ограниченно трудоспособными. Запрещаются контакты с ионизирующими излучениями, СВЧ-полями и иными мутя генными факторами. При артериальной гипертензии больные подростки освобождаются от экзаменов.
После адреналэктомии им необходим особенно щадящий режим. Профилактические прививки производятся строго по показаниям.
Призыву в армию они не подлежат, а впервые заболевшие с военного учета снимаются. В военные учебные заведения такие больные подростки не принимаются, даже при полном выздоровлении.
ВРОЖДЕННЫЙАДРЕНОГЕНИТАЛЬНЫЙСИНДРОМ
Определение. Врождённый адреногенитальныи синдром — генетическое заболевание, обусловленное недостаточностью ферментных систем коры надпочечникасоснижениемсинтезаглюкокортикоидовиминералокортикоидов при одновременно повышенной продукции андрогенов.
Синонимы: врожденная дисфункция коры надпочечников, врожденная вирилизирующая гиперплазия коры надпочечников, адреногенитальныи синдром.
Распространение. Врожденный адреногениталь ный синдром — основная и наиболее распростра-
ненная в мире патология надпочечников у подростком 95 % его составляют лица с врожденным дефектом 21гидроксилазы. В мире средняя частота классической 21-гидроксилазной недостаточности — от 1:5000 до 1:20000 в популяции. Но уэскимосов, особенно— у девочек, она очень велика —до 1:490 новорождённых Частота гетерозиготного носительства аутосомно-ре-цессивных дефектов 21 - гидроксилазы повсеместно весьма значительна — от 1:35 до 1:76, а у эскимосов является практически
сплошной (Штолеке X., 1970).
Ряд форм синдрома часто поражает евреев — неклассический вариант 21-гидроксилазного дефекта встречается у евреев-ашкенази с частотой до 3 %, что в 10 раз выше среднеевропейских цифр. Дефицит 11- в-гидроксилазы часто встречается у марокканских евреев.
Этиология и патогенез. Гиперпродукция надпо-
чечниковых половых стероидов известна в разных формах и сопровождает различные болезни и синдромы. Наиболее характерными общими проявлениями надпочечникового гиперандрогенизма служат гирсутизм, олигоменорея, угри и вирилизация.
Необходимо оговорить, чтонадпочечниковый гиперандрогенизм может иметь разную этиологию, и ввес-

ти понятиеоразнообразии адреногенитальных синдромов.
Традиционное собирательное название для нарушений продукции половых гормонов надпочечниками —
адреногенитальные синдромы. Эта сборная группа включает приобретенные и врождённые формы.
Приобретенные нарушения половых функций и признаков на почве гиперпродукции адренокортикальных половых гормонов связаны с различными опухолями, вырабатывающими половые стероиды, а врождённые— с наследственными дефектами ферментов стероидогенеза. Приобретенные адреногенитальные синдромы у подростков очень редки. Опухоли, вырабатывающие половые стероиды, как правило, возникают или манифестируют у взрослых. Тем не менее краткая характеристика приобретенных адреногенитальных синдромов должна быть предпослана под-
робному обсуждению одной из главных проблем подростковой эндокринологии — врожденного адреногениталъного синдрома, так как манифеста-
ция подобных опухолей уподростков совсем неисключается.
Выше уже описывалась возможность адренокортикального гиперандрогенизма при гиперкортицизме вследствие болезни и синдрома Иценко-Кушинга, в
частности, факт существования глюкоандросте-
ром.
Описаны андростеромы (1 -3 % всех опухолей над-
почечников) и кортикоэстромы надпочечников (от-
дельные наблюдения).
При андростеромах, которые у детей и подростков, как правило, злокачественные, а у взрослых — нередко бывают и доброкачественными, отмечается автономная гиперпродукция андрогенов, связанная с соматической мутацией (мутациями) генов стероидогенных энзимов в опухолевом клоне.
Андростеромы иногда достигают очень значительных размеров (до 1,5 кг весом). У женщин андростерома вызывает быструю вирилизацию (гирсутизм, перераспределение жира по мужскому типу, огрубение голоса, облысение, атрофия молочных желез, олигоменорея и аменорея, гипертрофия клитора, рельефность мускулатуры и повышение физической работоспособности, изменение стереотипов поло-ро- левого поведения).
У детей и подростков обоего пола развивается преждевременное половое созревание (у юношей — изосексуального, а у девушек — гетеросексуального типа). Рост костей в длину рано останавливается из-за опережающегокостноговозраста, больные остаются
невысокими. Это дополняется местными объемными и общеонкологическими симптомами, которые очень важны для дифференциальной диагностики, так как сходная картина может иметь множество вненадпочечниковых и неопухолевых причин. У взрослых мужчин многие случаи небольших по размеру андростером остаются, вероятно, нераспознанными.
При кортикоэстромах, которые, напротив, хорошо распознаются именно у юношей и мужчин, имеет место продукция эстрогенов в мутантном опухолевом клоне адренокортикальных клеток. Эти опухоли—зло- качественные, инвазивные, феминизирующие.
У юношей-носителей кортикоэстромы отмечается гинекомастия, часто—гипотрофия тестикул, феминизация телосложения и стереотипов поведения. Многие глюкостеромы, андростеромы и карциномы надпочечников выделяют некоторое количество эстрогенов, но это маскируется секрецией андрогенов.
Фактически врожденный адреногенитальный синдром — группа генетических заболеваний. Врождённая форма адренокортикального гиперандрогенизма связана с несколькими разными аутосомно-рецессивны- ми наследственными дефектами ферментов стероидогенеза, при которых возникший метаболический блок благоприятствует синтезу андростероидов в ущерб про-
дукции кортизола, а иногда — и минералокортикои-
дов. Эти наследственные заболевания входят в собирательную группу, известную как «врождённая гиперплазия коры надпочечников» (см. выше рис. 3 и
рис. 45).
Под действием рецессивных генов поражается один из ферментов биосинтеза кортикостероидов, начиная с эмбрионального периода. Патогенез обусловлен нарушением продукции одногоили нескольких кортикостероидов, в результате происходит задержка их синтеза на стадии половых стероидов, почему почти все формы таких энзимопатий дают разную степень нарушений полового развития.
Вместе с тем из-за ингибирования продукции кортикостероидов часто выпадает сервомеханизм, ограничивающий продукцию кортиколиберина и АКТГ, и гипо- таламо-гипофизарный нейросекреторный комплекс насыщает организм больных адренокортикотропи-
ном.
Врождённая гиперплазия коры надпочечников
(или врождённый адреногенитальный синдром) из-за наибольшей значимости в подростковой практике и в силу своего сложного характера, складывающегося из мозаичного и разнообразного сочетания проявлений гипо- и гиперкортицизма, нуждается в отдельном описании.
129

Впервые этот синдром описан в 1865 г. Л. Кречио, но его наследственный характер распознали лишь в середине XX столетия. Идея о ферментативном блоке стероидогенеза как основе данного расстройства принадлежит Ф. Барттеру с соавт. (1951).
Существует целое семейство генных мутаций, блокирующих те или иные этапы стероидогенеза и вызывающих варианты данного синдрома.
К ним относятся: дефекты 21-гидроксилазы, 11-в-
гидроксилазы, 3-в-олдегидрогеназы, 17-гидроксила- зы, 17-редуктазы, а-редуктазы, 20,22-десмолазы и 17,20-десмолазы.
На схеме (рис. 25) показаны участки путей стероидогенеза, где действуют эти ферменты, и локализуются данные блоки.
Выше уже было сказано, что от 90 до 95% случаев врожденного адреногенитального синдрома связаны с различными мутациями, снижающими активность 21гидроксилазы. Так как ген этого фермента СYP21 в хромосоме6 находится пососедству с генами главного комплекса гистосовместимости, у заболевания отмечается неравновесное сцепление с некоторыми его гаплотипа-
ми (В5, В14 Вw47, Вw51, Вw60, DRl7).
При различных мутациях гена CYP21 (делеция, инверсии) могут возникать разные клинические варианты 21-гидроксилазной недостаточности, известные как
классическая форма (в 75 % случаев — вирилизующая и солътеряющая, в 25 % случаев — только вирилизующая), а также неклассическая форма (отлича-
ющаяся менее тяжёлыми проявлениями, в частности, поздним проявлением вирильногосиндрома).
Классическая недостаточность 21-гидроксилазы ведет к полному блоку превращений 17-а-оксипро-
гестерона в 11 -дезоксикортизол и прогестерона в
11-дезоксикортизол. Избыток этих метаболитов превращается в андростендион, что ведет к повы-
шенной выработке надпочечниковых андрогенов.
Уже внутриутробно присутствует гиперандрогения. В то же время эффективность синтеза минералокорти-
коидов и глюкокортикоидов страдает, что ведет к усилению функции кортиколипотрофов аденогипофиза и к нарастанию уровня АКТГ, стимулирующего рост коры надпочечников и андростероидогенез.
При выраженном дефиците альдостерона гипертрофируется юкстагломерулярный аппарат почек. Кора надпочечников резко гиперплазируется за счет клубочковой и сетчатой зон. Возникают микроузловая или диффузная формы гиперплазии. Внешне вид надпочечников напоминает кору больших полушарий головного мозга. Вес одного надпочечника при норме в 6-7 граммов может достигать 60 граммов!
В адренокортикоцитах изобилуют липидные включения и присутствует липофусцин. В тестикулах мужчин могут бытьгиперплазияи дажеопухоли, а яичники уженщин проявляют признаки вторичной гипотрофии из-за высокого уровня андрогенов.
Клинически проявляются два синдрома—гиперан-
дрогенизм и гипокортицизм, причем второй — преиму щественно в форме первичного гипоалъдостерониз ма. При этом синдроме уже с рождения отмечаются признаки псевдогермафродитизма. У девочек вирилизуются наружные гениталии (гипертрофия клитора, смыканиеполовых губ потипумошонки, формированиеуро генитального синуса, напоминающего фаллоидную уретру). Мальчики при рождении имеют нормальныегениталии. Так как надпочечниковые андрогены относятся к слабым и при избытке конкурируют с тестостерономза рецепторы, тоиногда умальчиков такжемогут быть аномалии строения наружных половых органовгипоспадияи двусторонный крипторхизм.
В дальнейшем следует преждевременное половое созревание (pubertatio рrаесох): у девочек — по гете-
росексуальномутипу, а умальчиков — поизосексуальному.
Неклассический вариант синдрома приводит к рождению детей с внешне нормальными половыми признаками, но происходит постнатальная вирилизация и гетеросексуальное преждевременное половое созревание у девочек. У мальчиков развивается преждевременное изосексуальное половое созревание: рост ускорен и рано заканчивается окостенением метаэпифизарных хрящей, костный возраст опережает паспортный, отмечаются макрогенитосомия, низкорослость и гипертрихоз.
От 65 до 75 % лиц с классическим дефицитом 21-гид роксилазы страдают явным врожденным гипокорти- цизмом-гипоалъдостеронизмом. Отмечается врож-
денная гиперпигментация кожи. Со 2-5-й недели внеутробной жизни развивается сольтеряющий синдром. Появляются слабость, полиурия, гипотония мышц, гипотензия, потеря веса, гиперкалиемия, гипонатриемия, К гипохлоремия с характерной ЭКГ, ацидоз, срыгивание и фонтанирующая рвота, позднее—тяга к солёной пище. При сольтеряющей форме смертность от острой надпочечниковой недостаточности на 1-2-м году жизни очень велика иприближается к40% (ПлотниковаЕ.В., 1989).
У остальных гипокортицизм носит скрытый характер и компенсируется гиперренинемией и гиперпродук-
цией АКТГ.
Стёртыеи лёгкиеформы врожденного адреногениталъного синдромаслужатосновной причиной гирсутизма и адренархе у девушек. Различные авторы, обследуяпациентоксгирсутизмом,выявилилаборатор-
130

ныепризнаки синдрома узначительных групп — от 1,5 до30% отвсехслучаев гирсутизма!Очевидно, чтогирсутизм — достаточный повод для углублённогообследования на наличие21 -гидроксилазной формы синдрома путем измерения сывороточного базального и АКТГ-стимулированного уровня стероидного предшественника — 17-гидроксипрогестерона.
В отличие от андростером, при врожденном ад-
реногенитальном синдроме гиперпродукция андро-
генов чувствительна к подавлению дексаметазоном. При введении этого стероида понижаются содержание андрогенов в крови и экскреция их метаболитов — 17-KC — с мочой.
Болезнь может быть распознана путем антенатальной диагностики (амниоцентез, анализ ДНК клеток биоптата ворсин хориона).
В случае пренатального диагноза назначение дек-
саметазона матери блокирует гиперпродукцию андрогенов и позволяет избежать псевдогермафродитизма, в дальнейшем гормонально-заместительная терапия необходима в течение всей жизни.
Больные с врожденным адреногениталъным синдромом входят в группу высокого риска развития острой надпочечниковой недостаточности.
Некоторыми особенностями характеризуются остальные формы синдрома, затрагивающие кору надпочечникови зависящиеотдругихферментативныхдефектов (табл. 4).
Таким образом, гиперандрогенизм при мозаичных расстройствахфункций корынадпочечниковимеется
не всегда. С выраженной гиперпродукцией андроге-
нов надпочечниками протекают лишь некоторые из них, сжатую характеристику которых мы приводим ниже.
•Дефицит 3-в~олдегидрогеназы (нарушается переход D5-прегненолона в прогестерон, накапливается дегидроизоандростерон). Так как это ан-
дрогенное соединение обладает лишь незначительной активностью, то у девушек развивается нерезко выраженный вирилизм, а из-за де-
фицита активных андрогенов надпочечников и
тестикул у юношей могут быть черты наружного гермафродитизма. Это выражается в неполной маскулинизации гениталий. Отмечаются врождённые крипторхизм и гипоспадия.
•Дефицит ll-в-гидроксилазы приводит к наибо-
лее проксимальному блоку стероидогенеза — на этапе между 11 -дезоксикортизолом (субстанция «S» Рейхштейна) и кортизолом, а также между
11 -дезоксикортикостероном и кортикостеро-
ном. Результатом этого, наряду с другими симптомами, является вирилизация у девушек и pubertatio ргаесох — у детей.
•Дефицит 21-гидроксилазы приводит к нарушению перехода прогестерона в дезоксикортизол, а 17-а-прогестерона — в 11 -дезоксикортикос-
терон. Как уже указывалось выше, это — наиболее частая причина весьма выраженного гиперандрогенизма у детей и подростков со всеми вытекающими из этого последствиями.
Таблица4
Редкиеформыврожденнойгиперплазиикорынадпочечников
Дефектный |
Стероиды, |
Дефицит- |
Соединения, |
Примечания |
энзим |
выделение |
ные гормоны |
появляющиеся |
|
|
которых с |
|
в избытке |
|
|
мочой растёт |
|
|
|
|
|
|
|
|
20,22-десмо- |
Нет |
Все |
Холестерин в коре |
Тотальный гипокортицизм, гипогонадизм, |
лаза(синдром |
|
стероидные |
надпочечников не |
врождённый мужской псевдогермафро- |
Прадера- |
|
|
переходит в |
дитизм |
Гартнера) |
|
|
прегненолон |
|
Зр-олдегидро- |
5-3в- |
Глюко- |
Дегидроизоандро- |
Тяжелый гипокортицизм и гипогонадизм; |
геназа |
стероиды |
кортикоиды, |
стерон (слабый |
врожденный наружный псевдогерма- |
|
|
альдостерон |
андроген, антаго- |
фродитизм у плодов обоего пола |
|
|
|
нист тестостерона) |
|
17-а- |
Прегнандиол, |
Андрогены, |
Кортикостерон, |
Половой инфантилизм у женщин, псевдо- |
гидроксилаза |
тетрагидро- |
эстрогены, |
Дезоксикортико- |
гермафродитизм у мужчин; гипертензия, |
|
кортикостерон |
кортизол, |
стерон |
задержка соли и воды, гипокалиемический |
|
|
альдостерон |
|
алкалоз |
11(3- |
Тетрагидродезок- |
Глюкокорти- |
Андрогены, 11- |
Женский псевдогермафродитизм, |
гидроксилаза |
сикортизол,тет- |
коиды, |
дезокси- |
гипертензия, задержка соли и воды, |
|
рагидродезокси- |
альдостерон |
кортикостерон |
гипокалиемический алкалоз. Уступает по |
|
кортикостерон |
|
|
частоте (1:100000) лишь дефектам 21- |
|
|
|
|
гидроксилазы. |
131

Некоторые разновидности нарушения биосинтеза половых стероидов проявляются в основном расстройством их конверсии в периферических тканях. Они будут рассматриваться в разделе, посвященном нарушению полового созревания подростков.
Ниже приводятся детальные данные о клинике, диагностике и лечении самого частого у подростков ва-
рианта врожденного адреногениталъного синдрома, обусловленного дефицитом 21 -гидроксилазы.
Клиника. Картину врожденного адреногенитального синдрома определяет как вид энзиматического дефекта, так и степень выраженности продукции кор-
тизола, минералокортикоидов и тестостерона.
При недостаточности 21 -гидроксилазы подростки обычно низкорослы, непропорционально сложены, с широкими плечами и выраженной мускулатурой. Развитие у девушек идет по гетеросексуальному типу, а у юношей — по изосексуальному. В итоге у обоих полов прогрессирует вирилизация. У девушек нет месячных. Матка, яичники и молочные железы не развиваются, имеется гипертрофия клитора, рост волос — по мужскому типу, голос низкий. У юношей, несмотря на явную маскулинизацию, при большом пенисе развивается гипоплазия яичек и азоспер-мия.
Осложнения. Маскулинизация девочек с рождения является поводом ошибочного присвоения им мужского пола. У мальчиков может развиться опухоль яичек — лейдигома, в дальнейшем они могут стать бесплодными. Выраженный дефицит минералокортикоидов (75 % случаев врожденного адреногенитального синдрома вследствие дефицита 21-гидро- ксилазы) приводит к потере соли и к развитию недостаточности коры надпочечников, нередко — даже с летальным исходом.
При преобладании вирильной формы синдрома частичная продукция минералокортикоидов более или менее компенсирует потерю натрия и хлоридов, но выраженный дефицит алъдостерона при преимущественно сольтеряющейформевсегдаприводиткгипонатриемии.
Классификация. По клинической картине выделя-
ют вирилъную и сольтеряющую формы синдрома вследствие врожденного дефекта 21-гидроксилазы. Возможны разные варианты течения.
Диагностика. Диагностика дефицита 21-гидрокси- лазы основана на задержке синтеза кортикостероидов на стадии 17-оксипрогестерона, продукция которого повышается в десятки раз.
Так как истинный и ложный гермафродитизм может иметь множество не надпочечниковых и не эндокринных причин (см. ниже), всеподобные случаи явля-
132
ются поводом для определения хромосомного пола в других дифференциально-диагностических исследований во избежание ошибок в установлении пола в даль нейшем.
Вкрови повышены уровни 17-оксипрогестерона
итестостерона, а при сольтеряющей форме синдрома выявляется гипонатриемия и гиперкалиемия, что мо жет стимулировать повышенную продукцию ренина В моче — высокая экскреция 17-КС.
При рентгеноскопии выявляется раннее обызвеств-лениехрящейребер, дажеушныхраковини сухожилиймышц.Спомощьюлучевыхметодов диагностикимож-ноустановить степень гиперплазии коры надпочечни-ков и даже ее опухолевое перерождение.
Гинеколог может обнаружитьгипоплазиюи дажеотсутствие матки, яичников и влагалища.
При пробе с дексаметазоном (2 мг каждые 6 чая сов в течениедвух суток—всего32таблетки)этотсин- тетический глюкокортикоид на 50 % и более снижает у подростков с врожденным адреногенитальным синдромом секрецию 17-оксипрогестерона и экскрецию с мочой 17-КС.
Критерии диагноза. Выраженная маскулинизация юношей в сочетании с низкорослостыю, макрогенитосомия с гипоплазией яичек при отсутствии признаков интракраниальной опухоли типичны для этого синдро-ма. Большой клитор, гипоплазия внутренних половых органов при явных симптомах маскулинизации харак-терны для врожденного адреногенитальногосиндрома у девушек.
Уобоих полов обнаруживаются раннее закрытие зон роста костей, высокий уровень в крови 17-оксипроге-
стерона и тестостерона, гиперэкскреция 17-КС с
мочой.
Исследование полового хроматина, признаки гиперплазии коры надпочечников, подтверждённые применением УЗИ, КТ или МРТ-методик, помогают диагностировать врожденный адреногенитальныи синдроме дефицитом 21 -гидроксилазы.
Пример диагноза. Врожденная гиперплазия коры надпочечников в результате дефицита 21 -гидроксила- зы. Синдром потери хлорида натрия. Преждевременное половое развитие. Гипоплазия тестикул. Азоспермия.
Дифференциальный диагноз. Дифференциальная диагностика проводится с истинным гермафроди-
тизмом у девушек, с опухолями яичек у юношей (при этом преждевременного полового развития не бывает),
Маскулинизирующая опухоль коры надпочечников у
девушек (злокачественный врожденный адреногенитальный синдром)вызываетвыраженнуювирили

зацию, повышение в крови дегидроэпиандростерона, андростендиона и тестостерона, повышенную продукцию 17-КС. Проба с дексаметазоном отрицатель-
ная.
Преждевременное половое созревание возможно также при синдроме Пелицци, вызванном интракраниальными опухолями эпифиза, продуцирующими хо-
рионический гоиадотропин.
Исходы заболевания и прогноз. При сольтеряющей форме врожденного адреногенитального синдрома смерть от острой надпочечниковой недостаточно-
сти может наступить вскорепослерождения.
С возрастом потеря соли снижается, так как чувствительность почек к минералокортикоидам повышается.
При своевременном лечении прогноз относительно благоприятный: у девушек появляется феминизация, формируются молочные железы, влагалище (!), возникают нормальные месячные, при этом даже возможна репродуктивность.
Уюношей нормализуется сперматогенез, исчезают вторичные опухоли яичек, при постоянной терапии есть надежда на репродуктивность.
Постоянная терапия глюкокортикоидами может привести к развитию хронической недостаточности коры надпочечников или, напротив, к ятрогенному синдрому Иценко-Кушинга, артериальной гипертензии, язвенной болезни, отекам, аденоматозуяичеки надпочечников, к иммунодефициту.
Удевушек с врожденным адреногенитальным синдромом постоянная психическая травматизация может быть поводом к суициду.
Лечение. Лечение синдрома проводится непрерывно
ипожизненно.
При сольтеряющей формев диете ограничивают или
полностью исключают продукты, богатые калием (виноград, изюм, курага), добавляют поваренную соль. Лечение начинают с дексаметазона (2 мг каждые 6 часов в течение 2 суток с последующим снижением дозы до0,5-1 мг в сутки), послечего переходят на постоянный прием преднизолона (10 мг в сутки) под контролем уровня 17-оксипрогестерона в крови и 17КС— в моче.
Нарушения минерального обмена компенсируют добавкой поваренной соли.
Адекватность терапии оценивается по темпам физического развития и динамики костного возраста, по состоянию гениталий, поналичию кушингоидных симптомов, по уровню продукции 17-гидроксипрогесте- ронав крови и экскреции17-КС в моче.
В последние годы при сольтеряющей форме синдрома наряду с глюкокортикоидами используют флюд-
рокортизон (кортинефф, флоринеф). При добавке
минералокортикоидов течение даже вирильной формы явно улучшается.
При выраженной маскулинизации у девушек целесообразно прибегать к косметическим операциям во избежание психотравм. При опухолях надпочечников необходимо хирургическое лечение.
Профилактика. Профилактика врожденного адреногенитального синдрома как генетического заболевания пока проблематична.
В замкнутых популяциях (эскимосы) и изолятах можно рекомендовать экзогамные браки.
Диспансеризация. Диспансерная группа Д-3. Подростки с врожденным адреногенитальным син-
дромом нуждаются в пожизненном наблюдении эндокринологом с непрерывной терапией глюкокортикоида-
ми, а также минералокортикоидами.
У девушек с врожденным адреногенитальным синдромом нельзя менять пол на мужской без попытки их лечения глюкокортикоидами, которое дает удивительный эффект.
Безусловное стремление к сохранению у них муж-
ского пола — признак невежества врача. В решении вопроса о смене пола должны участвовать психиатр, эндокринолог, психолог, сексопатолог, гинеколог и уролог.
При плохом контроле лечения тяжелые осложнения могут стать причиной инвалидизации. При ранней адекватной терапии трудоспособность сохраняется. После операции по поводу маскулинизирующей опухоли одного надпочечника глюкокортикоиды вскоре отменяют, так как второй надпочечник сам начинает функциони-
ровать (Тюльпаков А.Н., 1991).
Вопросы экспертизы. Группа здоровья определяется индивидуально, в зависимости от клиники и возможностей компенсирующей терапии глюкокортикоидами. Показана ЛФК.
Психику страдающих врожденным адреногенитальным синдромом подростков следует щадить.
Пожизненная терапия глюкокортикоидами (даже по достижении выраженного лечебного эффекта) является поводом к освобождению всех подростков с врожденным адреногенитальным синдромом от военной службы.
К приему в военно-учебные заведения они не допускаются, что требует тщательного медицинского освидетельствования и эндокринологического обследования, так как их внешняя гипермаскулинизация может быть обманчивой.
133

РЕДКИЕ ФОРМЫ ПАРЦИАЛЬНОГО |
ренин. Ренинпродуцирующие клетки не отвечают |
|||
ГИПОКОРТИЦИЗМАУ ПОДРОСТКОВ |
на стимуляторы, например, на АКТГ. |
ги- |
||
Некоторые другие наследственные дефекты стеро- |
• Послеоперационный |
гипоренинемический |
||
идогенеза встречаются в подростковой практике ис- |
поалъдостеронизм развивается после удаления |
|||
ключительно редко. |
алъдостеромы. |
|
|
|
Так, при дефиците 18-дегидрогеназы отмечена |
• Гипоальдостеронизм может быть ятрогенным, в |
|||
изолированная гипопродукция минералокортико-идов |
результате передозировки и последующей рез- |
|||
или первичный изолированный гипоальдосте-ронизм. |
кой отмены экзогенных минералокортикоидов |
|||
Он может быть и результатом наследственного |
(например, флудрокортизона) и солодки (см |
|||
дефекта биосинтеза альдостерона на этапе 18- |
выше). В этих случаях он развертывается в связи |
|||
гидроксилазы. Данные формы генетического блока |
с подавлением ренинообразования в предыдущую, |
|||
стероидогенеза, в отличие от вариантов врождённой |
гиперальдостероновую, фазу. |
син- |
||
гиперплазии, не сопровождаются адреногениталъ- |
• Псевдогипоальдостеронизмом называется |
|||
ным синдромом и имеют тенденцию к спонтанной |
дром резистентности почек и других мишеней аль- |
|||
ремиссии. Отметим, что сходные по картине приобре- |
достерона к действию минералокортикоидных |
|||
тенные изолированные нарушения биосинтеза альдо- |
гормонов. Это обусловлено дефектом минера- |
|||
стерона могут возникать в любом возрасте и быть |
локортикоидных рецепторов. На фоне проявле- |
|||
следствием действия блокирующих аутоантител к 18- |
ний кажущегося гипоальдостеронизма уровни |
|||
дегидрогеназе. Такой гипоальдостеронизм сочетается |
альдостерона и ренина высокие. При псевдоги- |
|||
с другими аутоиммунными эндокринопатиями. |
поальдостеронизме затруднено лечение минера |
|||
локортикоидами, и приходится корригировать со- |
||||
Дефект биосинтеза альдостерона вызывают так- |
||||
стояние больных введением хлорида натрия. |
||||
же некоторые препараты — спиронолактоны (аль- |
||||
Данное состояние надо отличать от первичного |
||||
дактон, верошпирон). |
||||
гипоальдостеронизма. |
|
|||
При всех формах первичного гипоальдостеронизма |
|
|||
• Наследственный |
псевдогипоальдостеро- |
|||
уровень ренина в крови высокий. |
||||
низм — синдром Чика-Перри — описан этими |
||||
В дифференциально-диагностическом плане надо |
||||
авторамив 1958г. Онпроявляетсязадержкойпси |
||||
помнить о существовании вторичного изолированно- |
||||
хомоторного развития детей, гипонатриемией, |
||||
го гипоальдостеронизма при гипофункции ренин-ан- |
||||
потерей хлоридов инатриясмочой иизбыткомв |
||||
гиотензиновой системы. При этом варианте синдрома |
||||
крови калия. Дефект при этом заболевании —ча- |
||||
уровень ренина в крови низкий. |
||||
стичный ипреодолевается высокими дозами ми- |
||||
Симптомы любого гипоальдостеронизма включа- |
||||
нералокортикоидов (Шрейбер В., 1987). |
|
|||
ют гиперкалиемию, метаболический ацидоз, артери- |
|
|||
|
|
|
||
альную гипотензию, гипонатриемию и гиповолемию. |
ОСТРАЯ НАДПОЧЕЧНИКОВАЯ |
|
||
В тяжелых случаях бывают обмороки, брадикардия |
|
|||
НЕДОСТАТОЧНОСТЬ |
|
|||
вплоть до полной блокады сердца и приступов Мор- |
Определение. Острая |
недостаточность надпочеч- |
||
ганьи-Эдамса-Стокса с судорогами и помрачением |
||||
ников — это угрожающее жизни состояние в результа- |
||||
сознания. |
||||
те быстро наступающих снижения или полного прекра- |
||||
Существует несколько характерных состояний, при- |
||||
щения всех функций коркового и мозгового вещества |
||||
водящих к гипоренинемическому вторичному гипо- |
надпочечников. |
|
|
|
альдостеронизму. |
Синонимы: острая адреналовая недостаточность, |
|||
• Почечный канальцевый ацидоз IV типа мало- |
||||
острый гипокортицизм, острый гипоадренализм, гипоад- |
||||
актуален для подростковой медицины, так как |
реналовый криз, аддисоновый криз, молниеносная пур- |
|||
поражает в основном пожилых мужчин. Но у под- |
||||
пура (purpura fulminans), синдром Уотерхауза-Фридерик- |
||||
ростков он может встретиться при тяжелых пред- |
сена. |
|
|
|
шествующих заболеваниях — подагре, инсулин- |
Распространенность. |
Острая недостаточность |
||
зависимом сахарном диабете, хроническом |
надпочечников может возникать в любом возрасте, |
|||
пиелонефрите. Этот синдром является следстви- |
но чаще — у детей и у рожениц. Половых различий в |
|||
ем прогрессирующего нефросклероза, охватываю- |
частоте патологии нет. У подростков наблюдается |
|||
щего юкстагломерулярный аппарат почек. Продук- |
редко, в основном при менингококковом или стрепто- |
|||
ция ренина падает, страдает переходпроренина в |
кокковом сепсисах. |
|
|
|
134

Этиология и патогенез. Острая недостаточность надпочечников (синдром Уотерхауза-Фридериксена) чаще всего представляет собой внезапное резкое снижение функции надпочечников вследствие разрушения их коркового и мозгового веществ. Наиболее часто она возникает в ходе синдрома диссеминированного внут-
рисосудистогсвёртывания-тромбообразования. Причиной тромбозов, инфарктов и апоплексии надпочечников служит активация тромбогенных свойств их эндотелия циркулирующими цитокинами при системном действии медиаторов воспаления, характерном длясепсисаитоксико-септическогошока.
Большое значение в генезе синдрома имеют генерализованные инфекции с бактериемией и вирусемией на фоне наследственных дефектов системы комплемента и других причин иммунодефицита. Наиболее часто синдром является осложнением менингококкового сепсиса. Отмечены его случаи также при стрептококковой инфекции, при гриппе и в дебюте тяжёлого полиомиелита, при гематогенном диссеминированном туберкулезе, а при резко сниженном иммунитете острое поражение надпочечников может вызывать генерализованная синегнойная и даже цитомегаловирусная инфекции.
Механизм поражения имеет много общего с экспериментально получаемой на животных аллергоидной (псевдоаллергической) реакцией на липополисахари-
ды— феноменом Санарелли-Шварцмана.
При этих состояниях эндотоксины бактерий и цитокины иммунной системы вызывают экспрессию дополнительных рецепторов цитокинов и молекул клеточной адгезии на поверхности эндотелия сосудов ряда органов, в том числе — надпочечников. При волнах бактериемии провоцируется активация сторожевой полисистемы плазмы, включая кинины и свёртывание, а также фибринолиз и комплемент, что и ведёт к региональному или системному ДВС-синдрому, захватывающему сосуды надпочечников. В результате надпочечники могут превращаться буквально в заполненные свернувшейся кровью мешочки.
Неифекционные факторы, способствующие синдрому —травма (в том числе, родовая), тяжелые длительные стрессы при продолжительных хирургических вмешательствах, ранениях, ожоговой болезни, родах. Синдром может осложнять геморрагические диатезы, острую и хроническую лучевую болезнь.
Проявления синдрома практически идентичны тяжёлому аддисонову кризу (см. ниже). Артериальная гипотензия и гипогликемия бывают по обыкновению очень сильно выраженными. На клиническую картину наслаиваются симптомы тромбо-геморрагического синдрома(например,петехииидажевыраженныеподкожные
кровоизлияния, что позволило назвать этот синдром
молниеносной пурпурой), а также симптомы основ-
ного заболевания (например, менингеальные явления, лихорадка и др.).
Для этой формы синдрома бывает достаточно ге- моррагическо-некротических изменений даже в одном надпочечнике при частичной сохранности второго.
Острая недостаточность надпочечников, сходная с классическим синдромом Уотерхауза-Фридериксена, может наступать и в результате адреналэктомии, при острой декомпенсации хронической недостаточности коры надпочечников, при внезапной отмене продолжительной глюкокортикоидной терапии.
При этом возникает острое нарушение процессов адаптации и всех видов обмена.
Острое прекращение продукции глюко- и минералокортикоидов приводит к внезапному снижению в крови уровней натрия, хлоридов, глюкозы, к угрожающему повышению концентрации калия, к падению артериального давления, коллапсуи шоку.
В результате развиваются ацидоз, выраженная гипогликемия, эксикоз, олигурия и анурия. Клиническая картина обычно развертывается быстро (за 1-2 часа), иногда — несколько медленнее (за сутки).
Клиника. Клиника может варьировать при различной этиологии. Возникают головная боль, боли в животе с тошнотой, рвотой, судороги.
Состояние сразу тяжелое — с интоксикацией и лихорадкой до 41,5 °С, одышкой, цианозом, возбуждением, бредом, реже — с адинамией и астенизацией. Кожа сухая, тургор ее понижен, появляются геморрагические высыпания звездчатого характера, которые обычно нарастают снизу вверх — конечности, мошонка, спина — с тенденцией к слиянию (рис. 49).
Возможны менингеальные признаки. Появляются симптомы гипогликемии. Развивается прострация, при этом артериальное давление вообще может не определяться. В результате коллапса и шока нередко наступает молниеносная смерть.
Осложнения. Течение заболевания всегда тяжелое или крайне тяжелое. При инфекционно-септической и травматической этиологии доминируют проявления системного действия медиаторов воспаления, обусловливающие шок.
Бывает кетоацидоз (при гипогликемии!). Могут развиться высочайшая гипертермия с дегидратацией, острая почечная недостаточность, необратимый коллапс, далее — смешанная по этиологии кома, что является причиной летальных исходов.
Классификация. Различают следующие клиническиеварианты острой недостаточности надпочечников:
135
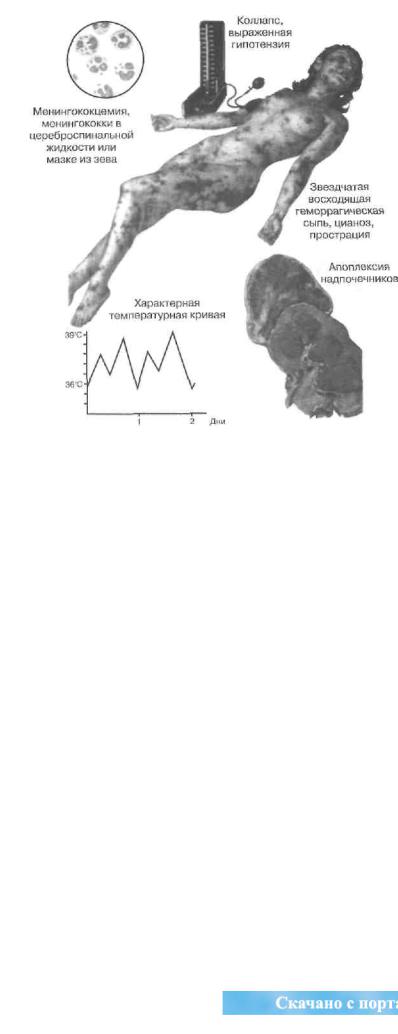
|
|
|
|
|
|
показателей калия, мочевины, креатинина, гематокрита |
|||
|
|
|
Рис. |
|
|
||||
49. |
|
|
|
|
|
и СОЭ. |
|||
|
|
|
|
|
|
В моче появляются белок, лейкоциты, эритроциты |
|||
|
|
|
|
|
|
||||
|
|
|
|
|
|
и цилиндры, кетоновые тела (несмотря на гипоглике- |
|||
|
|
|
|
|
|
мию). |
|||
|
|
|
|
|
|
ЭКГ отличается низким вольтажом, удлинением |
|
|
|
|
|
|
|
|
|
интервала QT, высокими и острыми зубцами Т. Арте- |
|
|
|
|
|
|
|
|
|
риальное давление всегда снижается в различной, но |
|
|
|
|
|
|
|
|
|
выраженной степени (вплоть до нуля), что всегда весь- |
|
|
|
|
|
|
|
|
|
ма подозрительно на острую патологию надпочечни- |
|
|
|
|
|
|
|
|
|
ков. |
|
|
|
Синдром |
|
|
|
||||||
|
|
|
|
Критерии диагноза. Общие клинические призна-ки |
|||||
Уотерхауза- |
|
|
|
|
|||||
|
|
этой патологии — острая сердечно-сосудистая не- |
|||||||
|
|
|
|
|
|
||||
|
|
|
|
|
|
достаточность, коллапс, абдоминальный синдром (без |
|||
|
|
|
|
|
|
симптомовраздражениябрюшины),геморрагии вкожуи |
|||
|
|
|
|
|
|
слизистые. |
|||
|
|
|
|
|
|
В диагностике ориентируются в основном на разви- |
|
|
|
|
|
|
|
|
|
тие коллапса и шока. Важно срочное исследование на- |
|
|
|
|
|
|
|
|
|
трия и калия в крови и определение их соотношения |
|
|
|
|
|
|
|
|
|
(величина Na/K падает до 20 и ниже), определение в |
|
|
|
|
|
|
|
|
|
крови уровня глюкозы (падает). |
|
|
|
|
|
|
|
|
|
Наблюдается типичная для гипокортицизма эози- |
|
|
|
|
|
|
|
|
|
нофилия (более 50 в 1 мкл крови). |
|||
|
|
|
|
|
|
При наличии технических возможностей необходи- |
|
||
|
|
|
|
|
|
мо срочное исследование крови на содержание корти- |
|
||
Фридериксена (поФ. Неттеру, СIВА) |
|
зола и альдостерона. Даже поздние результаты ана- |
|
||||||
|
—сердечно-сосудистый (с преобладанием артери- |
|
лизов крови на кортикостероиды очень важны для |
|
|||||
|
|
понимания причин гибели подростка при неясном ди- |
|
||||||
|
альной гипотонии, коллапса и шока); |
|
агнозе. |
|
|||||
|
—желудочно-кишечный (диспепсияи боли в живо- |
|
У подростков с аддисоновой болезнью в анамне-зе |
|
|||||
|
те); |
|
диагностика несравненно проще; острая недостаточ- |
||||||
|
—нервно-психический (бред, зрительныегаллюци- |
|
ность надпочечников при аддисоновом кризе (гипохло- |
||||||
|
нации, астения, депрессия). |
|
ремической коме) нарастает, к счастью, не так быстро, |
||||||
|
Диагностика. Отсутствие в рядовых медицинских |
|
с клиникой прекомы, что упрощает проведение диагно- |
||||||
учреждениях возможностей экстренного исследования |
|
стических процедур. |
|||||||
в крови кортикостероидов существенно затрудняет |
|
Пример диагноза. Эпидемический менингит. Me- |
|||||||
диагностику острой недостаточности надпочечников. |
|
нингококковый сепсис. Кровоизлияниев надпочечни- |
|||||||
Патогномоничных симптомов заболевания, кроме звёз- |
|
ки. Острая недостаточность надпочечников. Коллапс. |
|||||||
дчатой геморрагической сыпи на коже, нет (да и гемор- |
|
Геморрагии в области нижних конечностей, груди и |
|||||||
рагии в зависимости от этиологии синдрома наблюда- |
|
живота. |
|||||||
ются не всегда). |
|
Дифференциальный диагноз. Сходные симптомы |
|||||||
|
Всегда важен анамнез: предшествующие инфекции |
|
возможны при системных аллергоидных реакциях, |
||||||
или контакты с инфекциями (эпидемический менингит, |
|
вызванных разными суперантигенами. |
|||||||
грипп), травмы и ушибы (особенно поясничной облас- |
|
Поэтому исключают пищевую токсикоинфекцию, |
|||||||
ти), ожоги, затянувшиеся операции, облучение, хрони- |
|
синдром стафилококкового токсического шока, |
|||||||
ческая недостаточность коры надпочечников (аддисо- |
|
токсический вариант плевропневмонии, токсико- |
|||||||
нова болезнь). Нередко провоцирующий фактор |
|
дермии (синдром Лайелла-Стивенса-Джонсона), |
|||||||
установить невозможно. |
|
геморрагические диатезы (болезнь Шенляйн-Гено-ха, |
|||||||
|
В крови обнаруживаются умеренный нейтрофиль- |
|
тяжелая цинга). |
||||||
ный лейкоцитоз, эозинофилия, тромбоцитопения, сниже- |
|
Проводят дифференциальную диагностику кома- |
|||||||
ниеуровней натрияи хлоридов, глюкозы, повышение |
|
тозных состояний. |
|||||||
136 |
|
|
|
|
|
|
|
|
|