

- •Preface to the First Edition
- •Preface to the Second Edition
- •Contents
- •Diagnostic Challenges
- •Expert Centers
- •Patient Organizations
- •Clinical Trials
- •Research in Orphan Lung Diseases
- •Orphan Drugs
- •Orphanet
- •Empowerment of Patients
- •Conclusions
- •References
- •Introduction
- •Challenges to Overcome in Order to Undertake Quality Clinical Research
- •Lack of Reliable Data on Prevalence
- •Small Number of Patients
- •Identifying Causation/Disease Pathogenesis
- •Disease Complexity
- •Lack of Access to a Correct Diagnosis
- •Delay in Diagnosis
- •Challenges But Not Negativity
- •Some Success Stories
- •The Means to Overcome the Challenges of Clinical Research: Get Bigger Numbers of Well-Characterized Patients
- •The Importance of Patient Organizations
- •National and International Networks
- •End Points for Trials: Getting Them Right When Numbers Are Small and Change Is Modest
- •Orphan Drug Development
- •Importance of Referral Centers
- •Looking at the Future
- •The Arguments for Progress
- •Concluding Remarks
- •References
- •3: Chronic Bronchiolitis in Adults
- •Introduction
- •Cellular Bronchiolitis
- •Follicular Bronchiolitis
- •Respiratory Bronchiolitis
- •Airway-Centered Interstitial Fibrosis
- •Proliferative Bronchiolitis
- •Diagnosis
- •Chest Imaging Studies
- •Pulmonary Function Testing
- •Lung Biopsy
- •Mineral Dusts
- •Organic Dusts
- •Volatile Flavoring Agents
- •Infectious Causes of Bronchiolitis
- •Idiopathic Forms of Bronchiolitis
- •Connective Tissue Diseases
- •Organ Transplantation
- •Hematopoietic Stem Cell Transplantation
- •Drug-Induced Bronchiolitis
- •Treatment
- •Constrictive Bronchiolitis
- •Follicular Bronchiolitis
- •Airway-Centered Interstitial Fibrosis
- •Proliferative Bronchiolitis
- •References
- •Background and Epidemiology
- •Pathophysiology
- •Host Characteristics
- •Clinical Manifestations
- •Symptoms
- •Laboratory Evaluation
- •Skin Testing
- •Serum Precipitins
- •Eosinophil Count
- •Total Serum Immunoglobulin E Levels
- •Recombinant Antigens
- •Radiographic Imaging
- •Pulmonary Function Testing
- •Histology
- •Diagnostic Criteria
- •Historical Diagnostic Criteria
- •Rosenberg and Patterson Diagnostic Criteria
- •ISHAM Diagnostic Criteria
- •Cystic Fibrosis Foundation Diagnostic Criteria
- •General Diagnostic Recommendations
- •Allergic Aspergillus Sinusitis (AAS)
- •Natural History
- •Treatment
- •Corticosteroids
- •Antifungal Therapy
- •Monoclonal Antibodies
- •Monitoring for Treatment Response
- •Conclusions
- •References
- •5: Orphan Tracheopathies
- •Introduction
- •Anatomical Considerations
- •Clinical Presentation
- •Etiological Considerations
- •Idiopathic Subglottic Stenosis
- •Introduction
- •Clinical Features
- •Pulmonary Function Studies
- •Imaging Studies
- •Bronchoscopy
- •Treatment
- •Introduction and Clinical Presentation
- •Clinical Features
- •Pulmonary Function Studies
- •Imaging Studies
- •Bronchoscopy
- •Treatment
- •Tracheomalacia
- •Introduction
- •Clinical Features
- •Pulmonary Function Studies
- •Imaging Studies
- •Bronchoscopy
- •Treatment
- •Tracheobronchomegaly
- •Introduction
- •Clinical Features
- •Pathophysiology
- •Pulmonary Function Studies
- •Imaging Studies
- •Treatment
- •Tracheopathies Associated with Systemic Diseases
- •Relapsing Polychondritis
- •Introduction
- •Clinical Features
- •Laboratory Findings
- •Pulmonary Function and Imaging Studies
- •Treatment
- •Introduction
- •Clinical Features
- •Pulmonary Function Studies
- •Imaging Studies
- •Bronchoscopy
- •Treatment
- •Tracheobronchial Amyloidosis
- •Introduction
- •Clinical Features
- •Pulmonary Function Studies
- •Imaging Studies
- •Bronchoscopy
- •Treatment
- •Sarcoidosis
- •Introduction
- •Pulmonary Function Studies
- •Imaging Studies
- •Bronchoscopy
- •Treatment
- •Orphan Tracheopathies: Conclusions
- •References
- •6: Amyloidosis and the Lungs and Airways
- •Introduction
- •Diagnosis and Evaluation of Amyloidosis
- •Systemic AA Amyloidosis
- •Systemic AL Amyloidosis
- •Amyloidosis Localised to the Respiratory Tract
- •Laryngeal Amyloidosis
- •Tracheobronchial Amyloidosis
- •Parenchymal Pulmonary Amyloidosis
- •Pulmonary Amyloidosis Associated with Sjögren’s Disease
- •Conclusions
- •References
- •Introduction
- •Pathophysiology
- •Genetic Predisposition
- •Immune Dysregulation
- •Epidemiology
- •Incidence and Prevalence
- •Triggering Factors
- •Clinical Manifestations
- •General Symptoms
- •Pulmonary Manifestations
- •Ear, Nose, and Throat (ENT) Manifestations
- •Neurological Manifestations
- •Skin Manifestations
- •Cardiac Manifestations
- •Gastrointestinal Involvement
- •Renal Manifestations
- •Ophthalmological Manifestations
- •Complementary Investigations
- •Diagnosis
- •Diagnostic Criteria
- •Prognosis and Outcomes
- •Phenotypes According to the ANCA Status
- •Treatment
- •Therapeutic Strategies
- •Remission Induction
- •Maintenance Therapy
- •Other Treatments
- •Prevention of AEs
- •Conclusions
- •References
- •8: Granulomatosis with Polyangiitis
- •A Brief Historical Overview
- •Epidemiology
- •Pathogenesis
- •Clinical Manifestations
- •Constitutional Symptoms
- •Ear, Nose, and Throat (ENT) Manifestations
- •Pulmonary Manifestations
- •Kidney and Urological Manifestations
- •Kidney Manifestations
- •Urological Manifestations
- •Neurological Manifestations
- •Peripheral Nervous System (PNS) Manifestations
- •Central Nervous System (CNS) Manifestations
- •Spinal Cord and Cranial Nerve Involvement
- •Skin and Oral Mucosal Manifestations
- •Eye Manifestations
- •Cardiac Involvement
- •Gastrointestinal Manifestations
- •Gynecological and Obstetric Manifestations
- •Venous Thrombosis and Other Vascular Events
- •Other Manifestations
- •Pediatric GPA
- •Diagnosis
- •Diagnostic Approach
- •Laboratory Investigations
- •Biology
- •Immunology
- •Pathology
- •Treatment
- •Glucocorticoids
- •Cyclophosphamide
- •Rituximab
- •Other Current Induction Approaches
- •Other Treatments in GPA
- •Intravenous Immunoglobulins
- •Plasma Exchange
- •CTLA4-Ig (Abatacept)
- •Cotrimoxazole
- •Other Agents
- •Principles of Treatment for Relapsing and Refractory GPA
- •Outcomes and Prognostic Factors
- •Survival and Causes of Deaths
- •Relapse
- •Damage and Disease Burden on Quality of Life
- •Conclusions
- •References
- •9: Alveolar Hemorrhage
- •Introduction
- •Clinical Presentation
- •Diagnosis (Table 9.1, Fig. 9.3)
- •Pulmonary Capillaritis
- •Histology (Fig. 9.4)
- •Etiologies
- •ANCA-Associated Small Vessel Vasculitis: Granulomatosis with Polyangiitis (GPA)
- •ANCA-Associated Small Vessel Vasculitis: Microscopic Polyangiitis
- •Isolated Pulmonary Capillaritis
- •Systemic Lupus Erythematosus
- •Antiphospholipid Antibody Syndrome
- •Anti-Basement Membrane Antibody Disease (Goodpasture Syndrome)
- •Lung Allograft Rejection
- •Others
- •Bland Pulmonary Hemorrhage (Fig. 9.5)
- •Histology
- •Etiologies
- •Idiopathic Pulmonary Hemosiderosis
- •Drugs and Medications
- •Coagulopathy
- •Valvular Heart Disease and Left Ventricular Dysfunction
- •Other
- •Histology
- •Etiologies
- •Hematopoietic Stem Cell Transplantation (HSCT)
- •Cocaine Inhalation
- •Acute Exacerbation of Interstitial Lung Disease
- •Acute Interstitial Pneumonia
- •Acute Respiratory Distress Syndrome
- •Miscellaneous Causes
- •Etiologies
- •Pulmonary Capillary Hemangiomatosis
- •Treatment
- •Conclusions
- •References
- •Takayasu Arteritis
- •Epidemiology
- •Pathologic Features
- •Pathogenesis
- •Clinical Features
- •Laboratory Findings
- •Imaging Studies
- •Therapeutic Management
- •Prognosis
- •Behçet’s Disease
- •Epidemiology
- •Pathologic Features
- •Pathogenesis
- •Diagnostic Criteria
- •Clinical Features
- •Pulmonary Artery Aneurysm
- •Pulmonary Artery Thrombosis
- •Pulmonary Parenchymal Involvement
- •Laboratory Findings
- •Imaging Studies
- •Therapeutic Management
- •Treatment of PAA
- •Treatment of PAT
- •Prognosis
- •References
- •Introduction
- •Portopulmonary Hypertension (PoPH)
- •Epidemiology and Risk Factors
- •Molecular Pathogenesis
- •PoPH Treatment
- •Hepatopulmonary Syndrome (HPS)
- •Epidemiology and Risk Factors
- •Molecular Pathogenesis
- •HPS Treatment
- •Conclusion
- •References
- •12: Systemic Sclerosis and the Lung
- •Introduction
- •Risk factors for SSc-ILD
- •Genetic Associations
- •Clinical Presentation of SSc-ILD
- •Pulmonary Function Tests (PFTs)
- •Imaging
- •Management
- •References
- •13: Rheumatoid Arthritis and the Lungs
- •Introduction
- •Epidemiology
- •Risk Factors for ILD (Table 13.3)
- •Pathogenesis
- •Clinical Features and Diagnosis
- •Treatments
- •Prognosis
- •Epidemiology
- •Risk Factors
- •Clinical Features, Diagnosis, and Outcome
- •Subtypes or RA-AD
- •Obliterative Bronchiolitis
- •Bronchiectasis
- •COPD
- •Cricoarytenoid Involvement
- •Pleural Disease
- •Conclusion
- •References
- •Introduction
- •Systemic Lupus Erythematosus
- •Epidemiology
- •Pathophysiology
- •Pulmonary Manifestations
- •Pleural Disease
- •Shrinking Lung Syndrome
- •Thrombotic Manifestations
- •Interstitial Lung Disease
- •Other Pulmonary Manifestations
- •Prognosis
- •Sjögren’s Syndrome
- •Epidemiology
- •Pathophysiology
- •Pulmonary Manifestations
- •Airway Disorders
- •Lymphoproliferative Disease
- •Interstitial Lung Disease
- •Prognosis
- •Mixed Connective Tissue Disease
- •Epidemiology
- •Pathophysiology
- •Pulmonary Manifestations
- •Pulmonary Hypertension
- •Interstitial Lung Disease
- •Prognosis
- •Myositis
- •Epidemiology
- •Pathophysiology
- •Pulmonary Manifestations and Treatments
- •Interstitial Lung Disease
- •Respiratory Muscle Weakness
- •Other Pulmonary Manifestations
- •Prognosis
- •Other Therapeutic Options in CTD-ILD
- •Lung Transplantation
- •Conclusion
- •References
- •Introduction
- •Diagnostic Criteria
- •Controversies in the Diagnostic Criteria
- •Typical Clinical Features
- •Disease Progression and Prognosis
- •Summary
- •References
- •Introduction
- •Histiocytes and Dendritic Cells
- •Introduction
- •Cellular and Molecular Pathogenesis
- •Pathology
- •Clinical Presentation
- •Treatment and Prognosis
- •Erdheim-Chester Disease
- •Epidemiology
- •Cellular and Molecular Pathogenesis
- •Histopathology and Immunohistochemistry
- •Clinical Presentation
- •Investigation/Diagnosis
- •Chest Studies
- •Cardiovascular Imaging
- •CNS Imaging
- •Bone Radiography
- •Other Imaging Findings and Considerations
- •Disease Monitoring
- •Pathology
- •Management/Treatment
- •Prognosis
- •Rosai-Dorfman Destombes Disease
- •Epidemiology
- •Etiology/Pathophysiology
- •Histopathology and Immunohistochemistry
- •Clinical Presentation
- •Investigation/Diagnosis
- •Management/Treatment
- •Prognosis
- •Conclusions
- •Diagnostic Criteria for Primary Histiocytic Disorders of the Lung
- •References
- •17: Eosinophilic Pneumonia
- •Introduction
- •Eosinophil Biology
- •Physiologic and Immunologic Role of Eosinophils
- •Release of Mediators
- •Targeting the Eosinophil Cell Lineage
- •Historical Perspective
- •Clinical Presentation
- •Pathology
- •Diagnosis
- •Eosinophilic Lung Disease of Undetermined Cause
- •Idiopathic Chronic Eosinophilic Pneumonia
- •Clinical Features
- •Imaging
- •Laboratory Studies
- •Bronchoalveolar Lavage
- •Lung Function Tests
- •Treatment
- •Outcome and Perspectives
- •Clinical Features
- •Imaging
- •Laboratory Studies
- •Bronchoalveolar Lavage
- •Lung Function Tests
- •Lung Biopsy
- •Treatment and Prognosis
- •Eosinophilic Granulomatosis with Polyangiitis
- •History and Nomenclature
- •Pathology
- •Clinical Features
- •Imaging
- •Laboratory Studies
- •Pathogenesis
- •Diagnosis
- •Treatment and Prognosis
- •Long-Term Outcome
- •Hypereosinophilic Syndrome
- •Pathogenesis
- •Clinical and Imaging Features
- •Laboratory Studies
- •Treatment and Prognosis
- •Eosinophilic Pneumonias of Parasitic Origin
- •Tropical Eosinophilia [191]
- •Ascaris Pneumonia
- •Eosinophilic Pneumonia in Larva Migrans Syndrome
- •Strongyloides Stercoralis Infection
- •Eosinophilic Pneumonias in Other Infections
- •Allergic Bronchopulmonary Aspergillosis
- •Pathogenesis
- •Diagnostic Criteria
- •Biology
- •Imaging
- •Treatment
- •Bronchocentric Granulomatosis
- •Miscellaneous Lung Diseases with Associated Eosinophilia
- •References
- •Introduction
- •Pulmonary Langerhans’ Cell Histiocytosis
- •Epidemiology
- •Pathogenesis
- •Diagnosis
- •Clinical Features
- •Extrathoracic Lesions
- •Pulmonary Function Tests
- •Chest Radiography
- •High-Resolution Computed Tomography (HRCT)
- •Bronchoscopy and Bronchoalveolar Lavage (BAL)
- •Lung Biopsy
- •Pathology
- •Treatment
- •Course and Prognosis
- •Case Report I
- •Introduction
- •Epidemiology
- •Clinical Features
- •Histopathological Findings
- •Radiologic Findings
- •Prognosis and Therapy
- •Desquamative Interstitial Pneumonia
- •Epidemiologic and Clinical Features
- •Histopathological Findings
- •Radiological Findings
- •Prognosis and Therapy
- •Conclusion
- •References
- •19: Lymphangioleiomyomatosis
- •Introduction
- •Pathogenesis
- •Presentation
- •Prognosis
- •Management
- •General Measures
- •Parenchymal Lung Disease
- •Pleural Disease
- •Renal Angiomyolipoma
- •Abdominopelvic Lymphatic Disease
- •Pregnancy
- •Tuberous Sclerosis
- •Drug Treatment
- •Bronchodilators
- •mTOR Inhibitors
- •Anti-Oestrogen Therapy
- •Experimental Therapies
- •Interventions for Advanced Disease
- •Oxygen Therapy
- •Pulmonary Hypertension
- •References
- •20: Diffuse Cystic Lung Disease
- •Introduction
- •Lymphangioleiomyomatosis
- •Pathogenesis
- •Pathologic and Radiographic Characteristics
- •Diagnostic Approach
- •Pulmonary Langerhans Cell Histiocytosis (PLCH)
- •Pathogenesis
- •Pathological and Radiographic Characteristics
- •Diagnostic Approach
- •Birt-Hogg-Dubé Syndrome (BHD)
- •Pathogenesis
- •Pathological and Radiographic Characteristics
- •Diagnostic Approach
- •Lymphoproliferative Disorders
- •Pathogenesis
- •Pathological and Radiographic Characteristics
- •Diagnostic Approach
- •Amyloidosis
- •Light Chain Deposition Disease (LCDD)
- •Conclusion
- •References
- •Introduction
- •Lymphatic Development
- •Clinical Presentation of Lymphatic Disorders
- •Approaches to Diagnosis and Management of Congenital Lymphatic Anomalies
- •Generalized Lymphatic Anomaly
- •Etiopathogenesis
- •Clinical Presentation and Diagnosis
- •Course/Prognosis
- •Management
- •Kaposiform Lymphangiomatosis
- •Etiopathogenesis
- •Clinical Presentation and Diagnosis
- •Management
- •Course/Prognosis
- •Gorham Stout Disease
- •Etiopathogenesis
- •Clinical Presentation and Diagnosis
- •Management
- •Course/Prognosis
- •Channel-Type LM/Central Conducting LM
- •Etiopathogenesis
- •Clinical Presentation and Diagnosis
- •Management
- •Course/Prognosis
- •Yellow Nail Syndrome
- •Etiopathogenesis
- •Clinical Presentation and Diagnosis
- •Management
- •Course/Prognosis
- •Summary
- •References
- •Introduction
- •Historical Note
- •Epidemiology
- •Pathogenesis
- •Surfactant Homeostasis in PAP
- •GM-CSF Signaling Disruption
- •Myeloid Cell Dysfunction
- •GM-CSF Autoantibodies
- •Lymphocytosis
- •Clinical Manifestations
- •Clinical Presentation
- •Secondary Infections
- •Pulmonary Fibrosis
- •Diagnosis
- •Pulmonary Function Testing
- •Radiographic Assessment
- •Bronchoscopy and Bronchoalveolar Lavage
- •Laboratory Studies and Biomarkers
- •GM-CSF Autoantibodies
- •Genetic Testing
- •Lung Pathology
- •Diagnostic Approach to the Patient with PAP
- •Natural History and Prognosis
- •Treatment
- •Whole-Lung Lavage
- •Subcutaneous GM-CSF
- •Inhaled GM-CSF
- •Other Approaches
- •Conclusions and Future Directions
- •References
- •Introduction
- •Epidemiology
- •Gastric Contents
- •Pathobiology of GER/Microaspirate in the Lungs of Patients with IPF
- •GER and the Microbiome
- •Diagnosis
- •Clinical History/Physical Exam
- •Investigations
- •Esophageal Physiology
- •Upper Esophageal Sphincter
- •Esophagus and Peristalsis
- •Lower Esophageal Sphincter and Diaphragm
- •Esophageal pH and Impedance Testing
- •High Resolution Esophageal Manometry
- •Esophagram/Barium Swallow
- •Bronchoalveolar Lavage/Sputum: Biomarkers
- •Treatment
- •Anti-Acid Therapy (PPI/H2 Blocker)
- •GER and Acute Exacerbations of IPF
- •Suggested Approach
- •Summary and Future Directions
- •References
- •Introduction
- •Familial Interstitial Pneumonia
- •Telomere Related Genes
- •Genetic
- •Telomere Length
- •Pulmonary Involvement
- •Interstitial Lung Disease
- •Other Lung Disease
- •Hepatopulmonary Syndrome
- •Emphysema
- •Extrapulmonary Manifestations
- •Mucocutaneous Involvement
- •Hematological Involvement
- •Liver Involvement
- •Other Manifestations
- •Treatment
- •Telomerase Complex Agonists
- •Lung Transplantation
- •Surfactant Pathway
- •Surfactant Protein Genes
- •Pulmonary Involvement
- •Treatment
- •Heritable Forms of Pulmonary Fibrosis with Autoimmune Features
- •TMEM173
- •COPA
- •Pulmonary Alveolar Proteinosis
- •GMCSF Receptor Mutations
- •GATA2
- •MARS
- •Lysinuric Protein Intolerance
- •Lysosomal Diseases
- •Hermansky-Pudlak Syndrome
- •Lysosomal Storage Disorders
- •FAM111B, NDUFAF6, PEPD
- •Conclusion
- •References
- •Introduction
- •Pathophysiology
- •Clinical Presentation
- •Epidemiology
- •Genetic Causes of Bronchiectasis
- •Disorders of Mucociliary Clearance
- •Cystic Fibrosis
- •Primary Ciliary Dyskinesia
- •Other Ciliopathies
- •X-Linked Agammaglobulinemia
- •Chronic Granulomatous Disease and Other Disorders of Neutrophil Function
- •Other Genetic Disorders Predisposing to Bronchiectasis
- •Idiopathic Bronchiectasis
- •Diagnosis of Bronchiectasis
- •Management of Patients with Bronchiectasis
- •Airway Clearance Therapy (ACT)
- •Management of Infections
- •Immune Therapy
- •Surgery
- •Novel Therapies for Managing Cystic Fibrosis
- •Summary
- •References
- •Pulmonary Arteriovenous Malformations
- •Background Pulmonary AVMs
- •Anatomy Pulmonary AVMs
- •Clinical Presentation of Pulmonary AVMs
- •Screening Pulmonary AVMs
- •Treatment Pulmonary AVMs
- •Children with Hereditary Hemorrhagic Telangiectasia
- •Pulmonary Hypertension
- •Pulmonary Hypertension Secondary to Liver Vascular Malformations
- •Pulmonary Arterial Hypertension
- •Background HHT
- •Pathogenesis
- •References
- •27: Pulmonary Alveolar Microlithiasis
- •Introduction
- •Epidemiology
- •Pathogenesis
- •Clinical Features
- •Diagnosis
- •Management
- •Summary
- •References
- •Introduction
- •Hermansky-Pudlak Syndrome
- •Telomerase-Associated Pulmonary Fibrosis
- •Lysosomal Storage Diseases
- •Lysinuric Protein Intolerance
- •Familial Hypocalciuric Hypercalcemia
- •Surfactant Dysfunction Disorders
- •Concluding Remarks
- •References
- •Introduction
- •Background
- •Image Acquisition
- •Key Features of Fibrosis
- •Ancillary Features of Fibrosis
- •Other Imaging Findings in FLD
- •Probable UIP-IPF
- •Indeterminate
- •Alternative Diagnosis
- •UIP in Other Fibrosing Lung Diseases
- •Pleuroparenchymal Fibroelastosis (PPFE)
- •Combined Pulmonary Fibrosis and Emphysema
- •Chronic Hypersensitivity Pneumonitis
- •Other Fibrosing Lung Diseases
- •Fibrosing Sarcoidosis
- •CTD-ILD and Drug-Induced FLD
- •Complications
- •Prognosis
- •Computer Analysis of CT Imaging
- •The Progressive Fibrotic Phenotype
- •Other Imaging Techniques
- •Conclusion
- •References
- •Introduction
- •Bronchoalveolar Lavage (BAL)
- •Technique
- •Interpretation
- •Transbronchial Biopsy (TBB)
- •Transbronchial Lung Cryobiopsy (TLCB)
- •References
- •Introduction
- •Overview of ILD Diagnosis
- •Clinical Assessment
- •Radiological Assessment
- •Laboratory Assessment
- •Integration of Individual Features
- •Multidisciplinary Discussion
- •Diagnostic Ontology
- •Conclusions
- •References
- •Introduction
- •Idiopathic Pulmonary Fibrosis
- •Chronic Hypersensitivity Pneumonitis
- •Connective Tissue Disease
- •Drug-Induced Lung Diseases
- •Radiation Pneumonitis
- •Asbestosis
- •Hermansky-Pudlak Syndrome
- •Risk Factors for Progression
- •Diagnosis
- •Pharmacological Management
- •Conclusions
- •References
- •Historical Perspective
- •Epidemiology and Etiologies
- •Tobacco Smoking and Male Sex
- •Genetic Predisposition
- •Systemic Diseases
- •Other Etiological Contexts
- •Clinical Manifestations
- •Pulmonary Function and Physiology
- •Imaging
- •Computed Tomography Characteristics and Patterns
- •Thick-Walled Large Cysts
- •Imaging Phenotypes
- •Pitfalls
- •Pathology
- •Diagnosis
- •CPFE Is a Syndrome
- •Biology
- •Complications and Outcome
- •Mortality
- •Pulmonary Hypertension
- •Lung Cancer
- •Acute Exacerbation of Pulmonary Fibrosis
- •Other Comorbidities and Complications
- •Management
- •General Measures and Treatment of Emphysema
- •Treatment of Pulmonary Fibrosis
- •Management of Pulmonary Hypertension
- •References
- •Acute Interstitial Pneumonia (AIP)
- •Epidemiology
- •Presentation
- •Diagnostic Evaluation
- •Radiology
- •Histopathology
- •Clinical Course
- •Treatment
- •Epidemiology
- •Presentation
- •Diagnostic Evaluation
- •Radiology
- •Histopathology
- •Clinical Course
- •Desquamative Interstitial Pneumonia (DIP)
- •Presentation
- •Diagnostic Evaluation
- •Radiology
- •Histopathology
- •Clinical Course
- •Treatment
- •Epidemiology
- •Presentation
- •Diagnostic Evaluation
- •Radiology
- •Histopathology
- •Clinical Course
- •Treatment
- •References
- •Organizing Pneumonias
- •Epidemiology
- •Pathogenesis
- •Clinical Features
- •Imaging
- •Multifocal Form
- •Isolated Nodular Form
- •Other Imaging Patterns
- •Histopathological Diagnosis of OP Pattern
- •Etiological Diagnosis of OP
- •Treatment
- •Clinical Course and Outcome
- •Severe Forms of OP with Respiratory Failure
- •Acute Fibrinous and Organizing Pneumonia
- •Granulomatous Organizing Pneumonia
- •Acute Interstitial Pneumonia
- •Epidemiology
- •Clinical Picture
- •Imaging
- •Histopathology
- •Diagnosis
- •Treatment
- •Outcome
- •References
- •36: Pleuroparenchymal Fibroelastosis
- •Introduction
- •Epidemiology
- •Clinical Manifestations
- •Laboratory Findings
- •Respiratory Function
- •Radiologic Features
- •Pathologic Features
- •Diagnosis
- •Treatment
- •Prognosis
- •Conclusions
- •References
- •Introduction
- •Acute Berylliosis
- •Chronic Beryllium Disease
- •Exposure
- •Epidemiology
- •Immunopathogenesis and Pathology
- •Genetics
- •Clinical Description and Natural History
- •Treatment and Monitoring
- •Indium–Tin Oxide-Lung Disease
- •Hard Metal Lung
- •Flock Worker’s Disease
- •Asbestosis
- •Nanoparticle Induced ILD
- •Flavoring-Induced Lung Disease
- •Silica-Induced Interstitial Lung Disease
- •Chronic Silicosis
- •Acute and Accelerated Silicosis
- •Chronic Obstructive Disease in CMDLD
- •Simple CMDLD
- •Complicated CMDLD
- •Conclusion
- •References
- •38: Unclassifiable Interstitial Lung Disease
- •Introduction
- •Diagnostic Scenarios
- •Epidemiology
- •Clinical Presentation
- •Diagnosis
- •Clinical Features
- •Radiology
- •Laboratory Investigations
- •Pathology
- •Conclusion
- •References
- •39: Lymphoproliferative Lung Disorders
- •Introduction
- •Nodular Lymphoid Hyperplasia
- •Lymphocytic Interstitial Pneumonia (LIP)
- •Follicular Bronchitis/Bronchiolitis
- •Castleman Disease
- •Primary Pulmonary Lymphomas
- •Primary Pulmonary MALT B Cell Lymphoma
- •Pulmonary Plasmacytoma
- •Follicular Lymphoma
- •Lymphomatoid Granulomatosis
- •Primary Pulmonary Hodgkin Lymphoma (PPHL)
- •Treatment
- •References
- •Introduction
- •Late-Onset Pulmonary Complications
- •Bronchiolitis Obliterans (BO)
- •Pathophysiology
- •Diagnosis
- •Management of BOS
- •Post-HSCT Organizing Pneumonia
- •Other Late-Onset NonInfectious Pulmonary Complications (LONIPCs)
- •Conclusion
- •References
- •Introduction
- •Pulmonary Hypertension Associated with Sarcoidosis (Group 5.2)
- •PH Associated with Pulmonary Langerhans Cell Histiocytosis (Group 5.2)
- •PH in Combined Pulmonary Fibrosis and Emphysema (Group 3.3)
- •PH Associated with Lymphangioleiomyomatosis (Group 3)
- •Hereditary Hemorrhagic Telangiectasia (Group 1.2)
- •Pulmonary Veno-Occlusive Disease (Group 1.5)
- •Small Patella Syndrome (Group 1.2)
- •Conclusion
- •References
- •Introduction
- •Epidemiology
- •Timing, Chronology, Delay Time
- •Route of Administration
- •Patterns of Involvement [3, 4]
- •Drugs and Agents Fallen Out of Favor
- •Drug-Induced Noncardiac Pulmonary Edema
- •Drug-Induced Cardiogenic Pulmonary Edema
- •The “Chemotherapy Lung”
- •Drug-Induced/Iatrogenic Alveolar Hemorrhage
- •Drugs
- •Superwarfarin Rodenticides
- •Transfusion Reactions: TACO–TRALI
- •Acute Eosinophilic Pneumonia
- •Acute Granulomatous Interstitial Lung Disease
- •Acute Organizing Pneumonia (OP), Bronchiolitis Obliterans Organizing Pneumonia (BOOP), or Acute Fibrinous Organizing Pneumonia (AFOP) Patterns
- •Acute Amiodarone-Induced Pulmonary Toxicity (AIPT)
- •Accelerated Pulmonary Fibrosis
- •Acute Exacerbation of Previously Known (Idiopathic) Pulmonary Fibrosis
- •Anaphylaxis
- •Acute Vasculopathy
- •Drug-Induced/Iatrogenic Airway Emergencies
- •Airway Obstruction as a Manifestation of Anaphylaxis
- •Drug-Induced Angioedema
- •Hematoma Around the Upper Airway
- •The “Pill Aspiration Syndrome”
- •Catastrophic Drug-Induced Bronchospasm
- •Peri-operative Emergencies (Table 42.8)
- •Other Rare Presentations
- •Pulmonary Nodules and Masses
- •Pleuroparenchymal Fibroelastosis
- •Late Radiation-Induced Injury
- •Chest Pain
- •Rebound Phenomenon
- •Recall Pneumonitis
- •Thoracic Bezoars: Gossipybomas
- •Respiratory Diseases Considered Idiopathic That May Be Drug-Induced (Table 42.4)
- •Eye Catchers
- •Conclusion
- •References
- •Cancer Mimics of Organizing Pneumonia
- •Lung Adenocarcinoma/Bronchioloalveolar Carcinoma
- •Primary Pulmonary Lymphoma
- •Cancer Mimics of Interstitial Lung Diseases
- •Lymphangitic Carcinomatosis
- •Epithelioid Hemangio-Endothelioma
- •Lymphomatoid Granulomatosis
- •Cystic Tumors
- •Cavitating Tumors
- •Intrathoracic Pseudotumors
- •Respiratory Papillomatosis
- •Pulmonary Langerhans Cell Histiocytosis
- •References
- •Index
18 |
Langerhans Cell Granulomatosis and Smoking-Related Interstitial Lung Diseases |
325 |
|
|
|
a |
b |
|
c
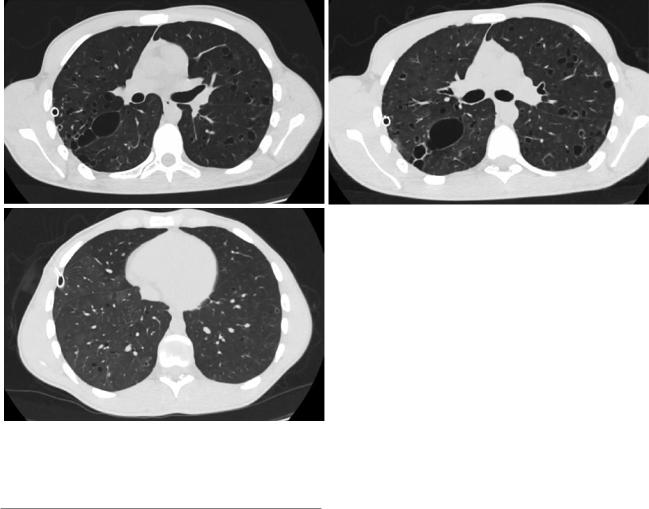
Fig. 18.6 (a) High-resolution CT scan demonstrates a small left pneumothorax with chest drainage and bilateral irregular cysts, with thin walls. (b) Some of these cysts appear to coalesce into larger and
irregular structures with bizarre shapes. (c) Image depicting the typical lesion distribution with relative sparing of lower lobes
Respiratory Bronchiolitis-associated
Interstitial Lung Disease (RB-ILD)
Introduction
Bronchiolitis is a generic term used to describe an infammatory process involving the small airways that may be the consequence of cigarette smoke exposure, infections, aspiration, environmental agents, drugs or underlying systemic disorders such as connective tissue diseases and transplantation rejection [102]. In 1974, Niewoehner described the presence of infammatory changes in the peripheral airways of a group of young smokers who had died of sudden death. The postmortem ndings showed the presence of clusters of pigmented macrophages in respiratory bronchioles and neighboring alveoli [103]. These changes, termed respiratory bronchiolitis (RB), is used to describe this form small airways infammation that occurs in virtually all smokers. In 1987, Myers et al. studied six smokers with clinical, functional, and radiological features suggestive of interstitial lung disease who underwent lung biopsy. The major pathologic ndings were the presence of respiratory bron-
chiolitis, characterized by clusters of pigmented alveolar macrophages within respiratory bronchioles, and, in addition, a mild chronic interstitial infammatory in ltrate of bronchiolar walls associated with hyperplasia of alveolar epithelial cells [104]. These were the rst cases described in literature of respiratory bronchiolitis associated with an infammatory and brotic involvement of the interstitium. The term “respiratory bronchiolitis with associated interstitial lung disease” (RB-ILD) appeared in 1989 in a study by Yousem et al. describing the histopathological differences between this new entity and other interstitial lung diseases. These and other studies, looking at the histopathological differences between RB and RB-ILD, have concluded that RB-ILD is usually associated with a greater extension ofbrosis, even though lungs of smokers affected by RB may also show mild alveolar brosis. The result is that differential diagnosis between RB and RB-ILD, exclusively based on histopathological and/or radiological ndings, can be very dif cult [105], and should also be based on clinical presentation. RB is usually asymptomatic while in RB-ILD symptoms such as dry cough and dyspnea are more often present [103].
326 |
C. Vancheri and S. Puglisi |
|
|
Cottin et al. studied a population of 79 smokers affected by spontaneous pneumothorax who underwent surgical biopsy. RB and RB-ILD were found in 88.6% and 67.1% of patients, respectively [106]. Emphysematous lesions were also present in a third of patients, collectively demonstrating the high incidence of these three pathologies in patients with spontaneous pneumothorax. However, explaining the mechanism by which tobacco induces change in small airways and the development of emphysema and bullae, remains an ongoing challenge.
The occurrence of RB is uncommon in the absence of smoke exposure. Fraig et al. studied a population of 109 patients affected by RB of whom 98% were smokers (current or former smokers). and only 2% were nonsmokers [107]. Woo et al. described a case of a nonsmoking woman with radiological and histological features of RB–ILD, who was continuously exposed to cigarette smoke because of her job. This study highlights the limitation of focusing on the incidence of RB in smokers and nonsmokers, as second-hand smoke and environmental exposure can also result in respiratory bronchiolitis [108].
BAL ndings in RB-ILD patients usually do not differ from those seen in normal “healthy” smokers and include an increased total number of cells or an increase in the percentage of macrophages that contain black tobacco pigment inclusions. A modest increase in neutrophils may also be present [109, 110].
Epidemiology
In a study performed on lung biopsy of smokers, Fraig et al. described the presence of RB in all smokers and about 50% of former smokers [107]. They also found an interesting correlation between the degree of pigmentation of macrophages and peribronchial brosis with the number of pack-years of cigarettes. In other studies, the relationship between RB and total smoke exposure is less obvious, although the percentage of smokers developing RB is consistently very high, ranging from 70% to 90%. RB-ILD is also closely related to smoking; different studies have shown that more than 90% of patients affected by RB-ILD are current smokers. RB-ILD typically affects smokers of 40–50 years of age with a slight male predominance and a history of cigarette smoking of 30 or more pack-year [5].
Clinical Features
The most common symptoms of RB-ILD are cough and dyspnea, and the typical physical exam ndings include inspiratory crackles and, more rarely, digital clubbing. Lung function tests are nonspeci c, and they can even be normal,
but more commonly will reveal obstructive, restrictive, or mixed obstructive/restrictive patterns. An obstructive pattern is a typical sign of smoking exposure and RB, while mixed patterns suggest a diagnosis of RB with interstitial lung disease . Diffusing capacity is usually decreased and may be a useful guide of severity of disease. DLCO values are not diagnostically useful, however, since they may be affected by other conditions related to cigarette smoking such as emphysema [42].
Histopathological Findings
According to Myers et al., who rst described the disease, RB-ILD and RB share many histological similarities [104]. The histological hallmark of both disorders is the accumulation of pigmented alveolar macrophages within respiratory bronchioles. RB macrophages are identi ed by abundant eosinophilic cytoplasm, with brown granular pigmentation representing constituents of cigarette smoke. A chronic infammatory cell in ltrate within bronchiolar walls is seen in RB-ILD, without honeycomb change or broblastic foci (Fig. 18.7). Myers and colleagues suggested that the main difference between RB and RB-ILD is based on the extent of the brosing and infammatory process, which in RB-ILD also involves adjacent alveolar walls in addition to airways [104]. Nakanishi et al. correlated the histopathological ndings of RB-ILD patients with radiological features. Accumulation of macrophages within bronchioles was associated with centrilobular micronodules (<3 mm), while the association of peribronchiolar infammation, brosis, and amassment of macrophages within the alveolar spaces corresponded to larger nodules (>3 mm). Ground glass opacities were the result of mild alveolar brosis accompanied by infammation and accumulation of macrophages. In more advanced stages of the disease, HRCT showed areas of linear and reticular opacity that histologically corresponded to alveolar and subpleural microcystic brosis [111]. Craig et al. compared the histological features of DIP and RB-ILD, studying their clinical and radiological correlation [112]. They studied 24 patients with RB-ILD and 25 with DIP. The typical histological feature of the two entities is the presence of intra-alveolar macrophages characterized by pigmented cytoplasm and associated with variable interstitial brosis and chronic infammation. RB-ILD lesions usually have a bronchiolocentric distribution, while DIP lesions are diffuse involving the entire pulmonary acinus. They also described a signi cantly greater extent of interstitial brosis, eosinophilic in ltration, and lymphoid follicle formation in DIP compared to RB-ILD. Yousem and coworkers examined nine cases of smokers with dyspnea, a mixed obstructive/restrictive pattern on lung function, reduced DLCO, and radiographic features of RB-ILD showing centrilobular nodules,
Данная книга находится в списке для перевода на русский язык сайта https://meduniver.com/
18 Langerhans Cell Granulomatosis and Smoking-Related Interstitial Lung Diseases |
327 |
|
|
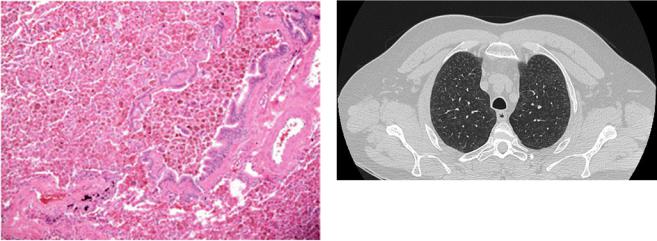
Fig. 18.7 RB consisting of distorted bronchioles with aggregates of “golden” macrophages into and around the bronchioles. (Courtesy of Professor G. Rossi. University of Modena-Reggio Emilia, Italy)
ground glass opacities, and emphysema. Surprisingly, lung biopsy revealed an extensive collagenous thickening of the alveolar septa with a patchy and subpleural distribution characteristic of nonspeci c interstitial pneumonia (NSIP). This study con rms how dif cult the differential diagnosis of these diseases may be, and highlights the critical role that smoking plays in brotic lung diseases [113].
Radiologic Findings
Chest X-ray is normal in up to 20–30% of cases of RB-ILD, and in other cases may show nonspeci c thickening of the central and peripheral bronchial walls, bilateral reticular- nodular opacities with diffuse distribution or upper lobe predominance [42]. The most common HRCT ndings are thickening of the bronchial walls, centrilobular nodules in the upper lung zones, and ground glass opacities (Fig. 18.8). Heyneman et al. compared the predominant HRCT features in a cohort of patients affected by RB, RB-ILD, and DIP, observing that in all of these conditions centrilobular nodules and ground glass opacities represent the most common radiological pattern [114]. In RB-ILD, these abnormalities are more profuse compared to RB and are characterized by the presence of areas of reticulation suggesting underlying interstitial brosis, albeit in the absence of honeycombing or traction bronchiectasis. There is no radiological cutoff that de nes the boundary between RB and RB-ILD. The radiological differential diagnosis may become even more complicated considering that HRCT ndings of RB-ILD may be very similar to those described for DIP [2]. DIP is characterized on CT scans by the presence of large areas of ground glass attenuation, which are usually bilateral, largely symmetrical, and peripheral and lower zone predominant. Basal,
Fig. 18.8 HRCT of a patient affected by RB-ILD showing poorly de ned centrilobular nodules
interlobular opacities can be associated with small peripheral cystic spaces with traction bronchiectasis. NSIP is marked by ground glass opacities, with irregular linear or reticular opacities and scattered micronodules in a subpleural distribution. In advanced disease, the presence of traction bronchiectasis and subpleural small cysts de ned as “microcystic honeycombing” may be helpful in making the correct diagnosis of NSIP [115].
Prognosis and Therapy
A number of studies that have linked smoking with the development of RB-ILD have demonstrated a clear improvement of the disease after smoking cessation. Nakanishi et al. have shown that smoking cessation alone, without any other treatment, leads to clinical, functional, and radiographic improvement. Symptoms and DLCO values were both improved after smoking cessation, as were ground glass opacities and centrilobular nodules on CT scans. A signi cant correlation between the change in DLCO and the reduction of centrilobular nodules and ground glass opacities was observed [111]. Sadikot et al., described two patients with biopsy-proven RB-ILD with dyspnea, severe lung function involvement, and respiratory failure who had signi cant clinical and functional improvement after smoking cessation [116]. Mavridou et al. reported a case of acute RB-ILD in which smoking cessation alone was not adequate, requiring steroid treatment. Gradual clinical, functional, and radiological improvement was reported, although it was necessary to continue high dose of corticosteroids for a longer period of time because of clinical deterioration following corticosteroid tapering [117]. Similarly, Woo et al. described a case of RB-ILD occurring in a patient exposed to second-hand cigarette smoke. The patient was treated with high dose of corticosteroids which resulted in improvement of ground glass opacities and centrilobular nodules on HRCT [108]. However, some other studies have not con rmed the effectiveness of smoking cessation and