

Эксперимент:
Таблица 1.9.1. Объем KMnO4 (V), пошедший на титрование и содержание окислителя
-
V(KMnO4), мл
M(K2Cr2O7), г
4.29
0.211
4.34
0.196
4.41
0.187
Заключение: (K2Cr2O7)=0.1990.0023(2.6%) Оценка выставлена удовлетворительно, так как число промахов равно 1.
2. Инструментальные методы анализа
Работа 2.1. Хроматография. Качественный анализ смесей предельных углеводородов
Цель работы: Установление наличия пентана, гексана, гептана, октана в смесях Х1 и Х2
Оборудование:
1. Газожидкостной лабораторный хроматограф ЛХМ-10
2. Микрошприц 10 мкл. 3. Секундомер.
Реактивы:
Эталонные вещества: гексан, гептан, октан, пентан.
Теория: Вид и основные параметры хроматограмм представлены на рис. 2.1.1 приложения к работе 2.1.
Качественные характеристики состава образцов:
1. tR – время удерживания компонента.
2. 0.5 – полуширина хроматографического сигнала.
3. Форма хроматографического сигнала (симметричная, асимметричная (фронт резкий или тыл резкий)).
Методика:
1. Получение хроматограмм. Поочередно микрошприцом отбираются 2 – 5 мкл эталонные и испытуемые образцы и вводятся прокалыванием прокладки в хроматограф. В этот момент включается секундомер. Не выключая секундомера производится измерение tR компонентов образца при наличии максимума хроматографического сигнала. Эксперимент заканчивается при получении сигнала от компонента имеющего наибольшее значение tR (в данной работе это октан).
2. Получение качественных характеристик.
Определение значения tR описано в п.1 методики. 0.5 определяется по хроматограмме в относительных единицах при помощи линейки (измеряется расстояние от базисной линии до максимума сигнала, это расстояние делится пополам и в точке половины высоты сигнала измеряется ширина сигнала, половина которого равна 0.5, см. также рис. 2.1.1). Форма сигнала устанавливается визуально (субъективно). Данные представлены в таблице 2.1.1.
3. Заключение о составе испытуемых образцов. На основании сравнения качественных характеристик эталонных и испытуемых образцов (данные представлены в табл. 2.1.1) делается вывод о составе Х1 Х2.
Эксперимент:
Таблица 2.1.1.Качественные характеристики эталонных и испытуемых образцов
|
Образец |
Время удерживания (tR), с |
μ0.5, о.е. |
Форма хроматографических сигналов |
|
Пентан |
13,5 |
2 |
Фронт резкий |
|
Гексан |
21,5; 21 |
3,1 |
Фронт резкий |
|
Гептан |
44; 41,5 |
5,5 |
Фронт резкий |
|
Октан |
63,5 |
7,9 |
Симметричный |
|
Смесь х1 |
15,5; 24,3 |
1,8; 2,9 |
|
|
Смесь х2 |
18,8; 34,5; 67 |
3; 4,5; 8,1 |
|
Заключение: Состав меси Х1 – пентан, гексан. Состав смеси Х2 – гексан, гептан, октан. (Результаты оценены на отлично).
Приложение к работе 2.1.
Теория:

Рис. 2.1.1. Хроматограмма двухкомпонентной смеси.
τ – время удерживания компонента; число теоретических тарелок N =5,54 (τ/0.5)2; высота условной теоретической тарелки Н = L/N, где L – длина хроматографической колонки.
Работа 2.2. Количественный анализ смеси предельных углеводородов
Цель работы: Определение процентной концентрации гексана и октана в смесях Х3 Х4 с показателем повторяемости 20%.
Оборудование: См. работу 1 (секундомер в данной работе не используется).
Реактивы: Эталонные смеси см. таб. 2.2.1.
Теория: Количественными характеристиками концентрации гексана и октана в объектах исследования являются высота или площадь хроматографического сигнала. Более предпочтительной является высота хроматографического сигнала из-за простоты ее измерения, а также предварительными экспериментами показана большая точность результатов при ее использовании. Для устранения из расчетов объема вводимой пробы (2-5мкл) используется не анализируемый компонент гептан, концентрация которого постоянна (30.0%). Этот компонент называется внутренним стандартом.
Методика:
1. Способ получения хроматограмм образцов указанных в таблице 2.2.1 описан в разделе методика п.1 работы 2.1.
2. Определение значения высоты хроматографического сигнала производится по хроматограмме в относительных единицах, например, при помощи линейки (измеряется расстояние от базисной линии до максимума сигнала). Эти данные представлены в виде h в таблице 2.2.1.
3. Расчет приведенных сигналов. Высоты сигналов гексана и октана делится на сигнал гептана (метод внутреннего стандарта), которые представлены в таблице 2.2.1 (h/).
4. Обработка данных. По программе СТ 5 рассчитываются градуировочные характеристики зависимости h/ гексана и октана от их концентрации. По h/ анализируемых компонентов в испытуемых образцах проводится расчет параметров указанных в цели работы и оценка результатов измерений.
Эксперимент:
Таблица 2.2.1. Состав эталонных растворов и сигналы от испытуемых растворов
|
Код образца |
Концентрации компонентов в образце, % | |||||||
|
Гексан |
Гептан |
Октан | ||||||
|
С, % |
hмм |
h/ |
C, % |
h |
С, % |
h |
h/ | |
|
1 |
10.0 |
8.0 |
0.31 |
30.0 |
26.1 |
60.0 |
31.6 |
1.21 |
|
2 |
20.0 |
19.5 |
0.56 |
30.0 |
34.8 |
50.0 |
36.9 |
1.06 |
|
3 |
30.0 |
18.1 |
0.67 |
30.0 |
32.0 |
40.0 |
32.2 |
0.92 |
|
4 |
40.0 |
39.8 |
0.91 |
30.0 |
36.8 |
30.0 |
26.7 |
0.73 |
|
5 |
50.0 |
69.9 |
1.04 |
30.0 |
67.3 |
20.0 |
33.8 |
0.50 |
|
X3 |
– |
12.2 |
0.79 |
30.0 |
15.5 |
– |
15.5 |
1.00 |
|
X4 |
– |
15.0 |
0.92 |
30.0 |
16.3 |
– |
10.5 |
0.64 |
Заключение: Х3: Cгексан = 35 ± 5 (15%), %, Соктан = 47 ± 5 (10%), %. Х4: Сгексан = 42± 5 (13%), %, Соктан = 26 ± 5 (19%), %
Поскольку достоверные значения концентрации равны: Х3 Cгексан = 25 %, Соктан = 45 %, Х4: Сгексан = 45 %, Соктан = 25 % , то результат анализа гексана в образце Х3 должен быть исключен из-за значимой систематической погрешности. Работа оценивается на удовлетворительно.
Приложение к работе 2.2.
Теория: Метод внутреннего стандарта: Внутренний стандарт используется для устранения из расчетов параметра (например, объем вводимой пробы) который невозможно точно измерить.
hi/=hi/hВС. Внутренний стандарт(не анализируемый компонент) вносится в одинаковых концентрациях в эталонные смеси и в испытуемые образцы.
Работа 2.3. Потенциометрия. Определение концентраций соляной HCl и уксусной кислот титрованием раствором NaOН
Цель работы:
1. Определение концентрации кислот по кривым титрования.
2. Оценка константы диссоциации CH3COOH.
3. Расчет погрешности измерения концентрации CH3COOH.
Оборудование:
1. Потенциометр рН 150.
2. Электроды: стеклянный и хлорсеребряный.
3. Бюретка 25.0 мл-3 шт.
Реактивы: Стандартный раствор NaOH; СNaOH = 0.102 моль/л.
Теория:
НCl
+ NaOH
![]() NaCl
+ Н2O
NaCl
+ Н2O
CH3COOH
+ NaOH
![]() CH3COONa
+ Н2O.
CH3COONa
+ Н2O.
СНА = СNaOH·VNaOH ТЭ /VНА (2.3.1),
KCH3COOH = [H+]·[CH3COO]/[CH3COOH] = 10–4.75 (25 oC) (2.3.2),
где НА – кислота, К – константа диссоциации.
Методика:
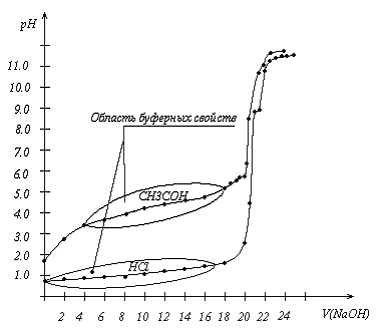
1. Потенциометрическое титрование. В стаканчик на 50 мл отбирается 20.0 мл кислоты. Далее в раствор помещаются электроды и измеряется рН раствора после стабилизации значения (примерно через 20 с). Из бюретки приливается по 0.50 мл раствора NaOH и измеряется рН раствора (титрование проводится до рН = 11). Данные представлены в таблице 2.3.1 и на рисунке 2.3.1.
2. Определение концентрации кислот. Графически измеряется объем NaOH в точке эквивалентности - VNaOH ТЭ (проводится перпендикуляр от точки перегиба на скачке титрования на ось VNaOH). Далее расчет проводится по формуле 2.3.1.
3. Определение константы диссоциации CH3COOH. Поскольку константа диссоциации для СН3СООН определяется по формуле 2.3.2, то в точке половинного оттитрования: KCH3COOH = [H+] и соответственно pН = pKCH3COOH. рН в точке половинного оттитрования находится проведением перпендикуляра от координаты VNaOH в точке VNaOH ТЭ/2 на кривую титрования и далее перпендикуляра на ось рН
4. Расчет погрешности измерений. В случае получения исполнителем только одной кривой титрования возможно объединение данных полученных другими исполнителями и провести расчеты погрешности по стандартной схеме указанной в приложении работы 2.2 (раздел теория).
Эксперимент: Экспериментальные данные по зависимости рН растворов кислот от объёма NaOH представлены на рисунке 2.3.1, а их численные значения в таблице 2.4.1 работы 2.4.
 ,
мл
,
мл
Рис. 2.3.1. Зависимость рН растворов кислот от объема титранта.
Заключение: СНСl = 0.10221.0/20.0 = 0.107 моль/л. ССН3СООН = 0.10220.8/20.0= 0.106 моль/л. Данные по ССН3СООН, полученные другими исполнителями: 0.108 и 0.104 моль/л таким образом:
ССН3СООН = 0.106 ± 0.005 (4.7%) моль/л.
рKCH3COOH = 4.3. Работа оценивается на отлично.
Приложение к лабораторной работе 2.3.
Теория: Потенциал стеклянного электрода: Е = Е0 + RT/(n F) lnan+= E0 + 0.059 рН, где Е0 – стандартный потенциал системы (В); Е – реальный потенциал; R – универсальная газовая постоянная, равная 8.3120 Дж/моль/К;.Т – абсолютная температура, К; F–постоянная Фарадея равная 96500 кл/моль; n–число электронов участвующих в реакции, или заряд потенциала определяемого иона; а – активность ионов Н+ (моль/л).
Работа 2.4. Потенциометрия. Использование метода Грана для измерения точки эквивалентности
Цель работы:
1. Измерение концентрации уксусной кислоты.
2. Оценка эффективности применения метода Грана по сравнению с методикой работы.
3. (В работе используются экспериментальные данные работы 3)
Теория:
F = VNaOH / 10pH; F/ = Fi / Fmax,
где F – функция Грана, а F/ - приведенная функция Грана, Fi – текущее значение функции Грана, Fmax – максимальное значение функции Грана (см. таблицу 2.4.1),значение рН при VNaOH равное нулю не используется.
Методика: Для расчета функции Грана используются экспериментальные данные рHCH3COOH = f(VNaOH) до точки эквивалентности и без данных при VNaOH = 0. Также не используются данные соответствующие началу титрования и не укладывающиеся в линейную зависимость (часто причиной этого является отсутствие настройки прибора).
Эксперимент:
Таблица 2.4.1. Зависимость функции Грана от объема NaOH
|
VNaOH, мл |
рHHCl |
рHCH3COOH |
F |
F/ |
|
0 |
0.76 |
1.82 |
|
|
|
2.0 |
0.80 |
2,92 |
0,002400 |
1.00 |
|
4.0 |
0.84 |
3,41 |
0,001560 |
0.650 |
|
6.0 |
0.89 |
3,70 |
0,001200 |
0.500 |
|
8.0 |
0.95 |
3,93 |
0,0009401 |
0.392 |
|
10.0 |
1.01 |
4.14 |
0,0007244 |
0.302 |
|
12.0 |
1.07 |
4.32 |
0,0005740 |
0.239 |
|
14.0 |
1.20 |
4.54 |
0,0003460 |
0.144 |
|
16.0 |
1.34 |
4.77 |
0,0002717 |
0.113 |
|
18.0 |
1.60 |
5.08 |
0,0001971 |
0.0821 |
|
18,5 |
|
5.20 |
0,0001176 |
0.0490 |
|
19.0 |
2.50 |
5.35 |
0,00008846 |
0.0368 |
|
19,5 |
5.42 |
5.47 |
0,00006607 |
0.0275 |
|
20.0 |
8.90 |
5.74 |
0,00003639 |
0.0151 |
|
20,5 |
9.99 |
6.44 |
0,000007443 |
0.00310 |
|
21.0 |
10.89 |
8.60 |
0,000005274 |
0.00129 |
Заключение: VNaOH ТЭ = 20.7±1.2(6.0%), мл
ССН3СООН = СNaOH·VNaOH ТЭ /VНА = 0.106 ± 0.007(6.0%)моль/л.
Случайная погрешность при определении точки эквивалентности методом Грана составляет приблизительно 6%. Использование метода Грана более экономично по сравнению с графическим методом нахождение точки эквивалентности (см. работу 3), поскольку для установления точки эквивалентности по методу Грана использованы 9 точек линейной зависимости F/ от VNaOH, что эквивалентно девяти кривым титрования. Однако, данную экономичность возможно реализовать только при наличии программного продукта для обработки экспериментальной зависимости VNaOH от рHCH3COOH. Работа оценивается на удовлетворительно вследствие завышенного значения доверительного интервала (обычное значение 3%).
Работа 2.5. Ионометрия
Цель работы: Измерение концентрации фторидов в электролите химического оксидирования в моль/л с показателем повторяемости 100%.
Оборудование:
1. Потенциометр рН – 340.
Электроды: хлорсеребряный и лантанфторидный.
Пипетки – 1.00мл и 5.00мл, мерная колба = 50.0 мл, мерный цилиндр – 10мл.
Емкости для растворов NaF (№2, №3, №4, №5).
Реактивы:
1. Стандартный раствор NaF (CF‾ = 1.00·10-1моль/л).
2. Ацетатный буферный раствор.
Теория: Е = E0 + S*pF, S = ∆E/∆рF,
где S – наклон линейной функции Е = f(pF), рF = -lgCF‾
Методика:
1. Приготовление растворов. Путем последовательного разбавления пипеткой отбирается 5.0 мл стандартного раствора NaF в мерную колбу. Далее в нее мерным цилиндром добавляется 10 мл ацетатного буфера и уровень раствора доводится дистиллированной водой до метки. Этот раствор перемешивается и переливается в емкость №2 (CNaF = 1.00*10-2 моль/л). Аналогично готовятся растворы NaF, указанные в таблице 1 (п. 3, 4, 5), при этом 5.0 мл раствора отбирается из предыдущей емкости. Далее пипеткой отбирается 1.00 мл испытуемого раствора в мерную колбу, после чего процесс приготовления раствора аналогичен описанному выше.
2. Измерение потенциала. В каждом из приготовленных растворов измеряется потенциал фторидселективного электрода относительно хлорсеребряного электрода, данные представлены в таблице 2.5.1.
Эксперимент:
Таблица 2.5.1. Зависимость потенциала фторидселективного электрода (Е) от концентрации F– (pF)
-
рF
E, мB
1.00
2.00
3.00
4.00
5.00
х
282
246
195
142
88
183
Заключение: рF = (3.2 ± 0.3 (9.8%)) моль/л. CF‾ = (9.5 ± 3.1 (30%))*10-4 моль/л. S = 49.2 мВ. Работа оценивается на отлично, поскольку достоверное значение CNaF = 1.00*10-3 моль/л.
Приложение к работе 2.5
Теория:
Схема гальванической цепи

Работа 2.6. Фотоколориметрия. Определение Сu(II)
Цель работы: Измерение концентрации Сu(II) в виде титра с показателем повторяемости 5%.
Оборудование:
1.Фотоэлектроколориметр КФК-2;
2. Кюветы 3.00мп 2шт; 3. Бюретка 25.0мл – 3шт;
4. Мерная колба 50.0мл.
Реактивы:
1.Стандартный раствор Сu(II) - CCu(II)эт =1,04*10–3 г/мл.
2. Раствор NН4ОН (1:1).
Теория:
Cu2+ + 2NH3*H2O = [Cu(ОН)2(NH3)2] + 2H+
A=*C*l,
где А- оптическая плотность, -молярный коэффициент светопоглощения (экстинкция) (л*моль-1*см-1), С – концентрация вещества, моль/л, L – толщина поглощения слоя, см.
Концентрации эталонных растворов, представленных в таблице, 2.6.1 рассчитываются по формуле СCu(II) = CCu(II)эт*Vэт/Vx,
Методика:
1. Приготовления растворов. В мерную колбу с помощью бюретки вводится определенный объем стандартного или испытуемого раствора соли меди Сu(II) (см. таблицу 2.6.1), прибавляется 5,0 мл раствора аммиака и доводится дистиллированной водой до метки.
2. Измерение оптической плотности растворов Сu(II). В качестве раствора сравнения используют дистиллированную воду. В две кюветы наливается дистиллированная вода после чего они помещаются в прибор. Показания оптической плотности устанавливают на нуль, после этого вода из одной кюветы выливается и в эту кювету наливается раствор Сu(II) и производится измерение оптической плотности (данные вносятся в таблицу 2.6.1).
3. Обработка данных. С помощью программы СТ - 5 рассчитываются метрологические характеристики градуировочной функции A = f(CCu(II)) и вычисляется значение концентрации Cu(II) в испытуемом образце и ее погрешность, а также выставляется оценка.
Эксперимент:
Таблица 2.6.1. Зависимость оптической плотности растворов от концентраций Cu(II)
|
VCu(II)эт, мл |
СCu(II)*103, г/мл |
А |
|
5.00 |
0.65 |
0,147 |
|
6.00 |
0.78 |
0,172 |
|
7.00 |
0.91 |
0,189 |
|
8.00 |
1.04 |
0,210 |
|
9.00 |
1.17 |
0,228 |
|
VCu(II)Х, мл |
СCu(II)Х, г/мл |
АХ |
|
8.00 |
1.04 |
0,165 |
Заключение: СCu(II) = (0.76 ± 0.02 (3.1%))•10-3 г/мл. Оценка хорошо, поскольку систематическая и случайная погрешности незначимы, но на градуировочной функции имеется один промах (0.78 – 0.172).
Приложение к работе 2.6.
Теория: Целью химического анализа является измерение химического состава или химической структуры объектов (параметры объектов).
Количество вещества измеряется: 1. измерением массы; 2. измерением объема; 3 измерением числа молей вещества (количество вещества кратное количеству элементарных единиц вещества = 6.02*1023 шт).
Концентрация вещества в объекте: количество вещества в стандартизованном количестве объекта (необходимо знать размерности концентрации – моль/л, г/мл, г/100г).
Содержание вещества в объекте: это количество вещества в объекте в целом.
Содержание совпадает с концентрацией когда объект имеет стандартные размеры.
Методика:
Рабочая область значений оптической плотности равна: А = 0.005 ÷ 0.800
Работа 2.7. Спектрофотометрия. Определение концентраций метилового оранжевого
Цель работы: Измерение концентрации метилового оранжевого (МО) в моль/л с показателем повторяемости 5%.
Оборудование:
1. Спектрофотометр СФ – 27.
2. Кювета 1.00 см (2 шт), мерная колба 25.0мл.
3. Бюретка 25.0мл (2 шт).
Реактивы: 1. Стандартный раствор метилового оранжевого (СМО СТ = 3.24*10-5 моль/л). 2. Буферный раствор рН = 1.68.
Теория: Закон светопоглощения описан в работе 6.

СМО = СМО СТ*VМО/ VМО Х (для расчета СМО в таблице 7.1)
Методика:
1. Приготовление раствора МО. В мерную колбу бюреткой отбирается объем МО указанный в таблице 2.7.1, далее раствор доводится до метки буферным раствором. Далее раствор МО наливается в кювету. В другую кювету наливается буферный раствор (раствор сравнения).
2. Измерение спектра поглощения раствора МО проводится в интервале 380 – 700 нм с шагом 20 нм. Область Аmax обнаруживается с шагом 5нм. Порядок измерения А: 1) установка длины волны; 2) установка раствора сравнения; 3) установка ручки щели около нуля; 4) открытие фотоэлемента; 5) вращением ручки щели установка показания прибора на нуль (А = 0); 6) установка раствора МО и снятие показания оптической плотности раствора МО (данные представлены на рисунке 2.7.1).
3. Измерение оптической плотности растворов МО приготовленных по п. 1 проводится при длине волны Аmax, найденной в п. 2 методики и далее измерение проводится аналогично п.2 (данные представлены в таблице 2.7.1).
4. Обработка данных. С помощью программы СТ - 5 рассчитываются метрологические характеристики градуировочной функции A = f(CМО) и вычисляется значение концентрации МО в испытуемом образце и ее погрешность, а также выставляется оценка.
Эксперимент:
Таблица 2.7.1. Зависимость оптической плотности раствора от концентраций метилового оранжевого
|
VМО, мл |
СМО* 105 гр/мл |
А |
|
3,0 4,0 5,0 6,0 7,0 6,0 (VМО Х) |
1,62 2,16 2,70 3,24 3,78 – |
0,584 0,679 0,890 1,05 1,26 0,84 |

Рис. 2.7.1. Зависимость оптической плотности растворов МО от длины волны излучения
Заключение: СМО = 2.5 ± 0.3(10%)∙10-5 г/мл.
Результат оценивается на неудовлетворительно вследствие занижения значения нормы погрешности в цели работы, а также завышения значений концентраций МО, что приводит к недопустимо высоким значениям оптической плотности растворов МО (больше 0.8). Необходимо уменьшить концентрацию стандартного раствора МО на порядок и увеличить объем проб растворов МО в три раза.
Работа 2.8. Кинетический метод анализа. Определение концентрации Мо(VI)
Цель работы: Измерение концентрации Мо(VI) в виде титра с показателем повторяемости 20%.
Оборудование:
1. Фотоэлектроколориметр.
2. Мерная колба 50.0мл.
3. Бюретка 25.0мл (3 шт).
4. Пипетка 2.0мл (2 шт)
5. Мерный цилиндр 10 мл.
6.Секундомер.
Реактивы:
1. Стандартный раствор (NH4)2MoO4 СМо(VI)СТ = 9.7*10–7 г/мл. 2. HCl (1:1).
3. Раствор KJ 0.20 моль/л.
4. Н2О2 0.20моль/л.
5. Крахмал – 0.3%.
Теория:
Н2О2 + 2J‾ + 2Н+ = J2 + 2Н2О.
Данная реакция (накопление йода в растворе) зависит от концентрации Мо(VI). Концентрация йода в растворе определяется фотоколориметрически по интенсивности посинения реакционного раствора в присутствии крахмала. Таким образом, индикаторным веществом является йод, а на фотоколориметре измеряется оптическая плотность раствора йодокрахмального комплекса. Причем реакцию, скорость которой определяется концентрацией анализируемого вещества, называют индикаторной.
Методика:
1. Приготовление растворов. В мерную колбу вводится стандартный или испытуемый раствор Мо(VI) объемом указанным в таблице 2.8.1 (столбец 2). Далее в колбу вводится 8.0 мл крахмала, 1.5 мл KJ, 20.0 мл HCl, до метки дистиллят. Далее в мерную колбу сверх метки добавляется 1.0 мл Н2О2 и тщательно все перемешивается в этот момент включается секундомер. Расчет концентрации стандартных растворов проводится по уравнению СМо(VI) = СМо(VI)СТ*VМо(VI)СТ/VМо(VI)Х.
2. Измерение оптической плотности растворов в ходе реакции производится аналогично работе 2.6 (раздел методика) при наступлении времени от начала реакции (время указано в таблице 2.8.1).
3. Обработка данных. С помощью программы СТ - 5 рассчитывается относительная скорость реакции накопления йода соответствующая углу наклона функции A = f(τ) (данные представлены в таблице 2.8.1, параметр VR). Далее вторично по программе СТ - 5 рассчитываются градуировочные характеристики зависимости VR от СMo(VI) и вычисляются параметры согласно цели работы, а также автоматически выставляется оценка.
Эксперимент:
Таблица 2.8.1. Зависимость оптической плотности реакционных растворов от концентрации Мо(VI) и времени реакции
|
№ n/n |
VMo(VI), Мл |
СMo(VI), г/мл |
A=f(τ). τ, с |
VR | ||||
|
90 |
120 |
150 |
180 |
210 | ||||
|
1 2 3 Образец Х |
2.0 5.0 8.0 8.0 |
2.433 6.08 9.730 – |
0.038 0.082 0.181 0.101 |
0.062 0.123 0.243 0.154 |
0.086 0.157 0.304 0.204 |
0.113 0.193 0.364 0.254 |
0.132 0.234 0.414 0.287 |
0.797 1.25 1.96 1.57 |
Заключение: СМо(VI) = (7.0 ± 1.3(18%))10-7 г/мл. Оценка работы – отлично.
Работа 2.9. Рентгенофлуоресцентный анализ (РФА). Анализ сплава на основе золота
Цель работы: Оценка пробы на золото образца сплава.
Оборудование:
Прибор VRA 20 L.
Банк спектров проб золота со значением 375, 583, 720.
Теория: Рентгеновское излучение (РИ) представляет собой электромагнитное излучение с длиной волны менее 100 нм. Для РФА используется РИ с длиной волны 0.10 – 2.0 нм.
Переход электрона в атоме с одного заполненного уровня на другой свободный уровень, имеющие одинаковые квантовые числа (например, переход со второго уровня на первый ближний к ядру атома – К серия) сопровождается испусканием характеристического РИ. Длина волны этого излучения для различных элементов описывается уравнением Мозли, которое является основой качественного элементного анализа:
1/λ = (АΖ - В)2 ,
где λ – длина волны характеристического РИ; Ζ – заряд ядра атома; А, В – постоянные величины.
В основе количественного анализа в первом приближении лежит линейная зависимость интенсивности характеристического РИ от концентрации элемента в пробе.
Методика:
1. Включается система водяного охлаждения.
2. Режим работы прибора: рентгеновская трубка с анодом из вольфрама, напряжением питания трубки 55 кВ, ток 30 мА кристалл–анализатор LiF 200, режим дискриминации излучения с уровнем 0.5 В; время релаксации τ = 1.
3. Образец помещается в пробоотборник и отправляется в спектральную камеру.
4. Проводится сканирование образца при углах 8 – 46 градусов ().
Заключение: По отношениям интенсивностей сигналов олова, серебра, меди, цинка и золота (сравнением со спектрами из банка спектров) сделано заключение о том, что образец имеет пробу 583.
Приложение к работе 2.9.
Теория: Выделенное с помощью коллиматора параллельное рентгеновское излучение падает на монокристалл где при определенном угле расположения монокристалла отражаются фотоны с только определенной длиной волны. Этот процесс описывается уравнением Вульфа – Брега:
n λ = 2 d Sin,
где d – межплоскостное расстояние монокристалла в нм.
Источником РИ является рентгеновская трубка. Прибор VRA 20L является спектрометром.
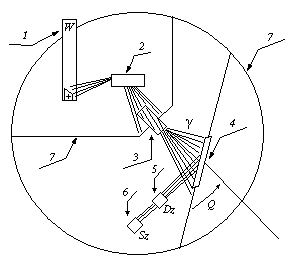
Рисунок 2.9.1. Принципиальная схема прибора VRA 20L
1 – рентгеновская трубка. 2 – анализируемый образец. 3 – коллиматор. 4 – монокристалл. 5 – проточный детектор.
6 – сцинтилляционный детектор. 7 – защитный кран.