

книги / Получение наночастиц и наноматериалов
..pdfГЛАВА 2. СУПРАМОЛЕКУЛЯРНЫЕ АНСАМБЛИ И УСТРОЙСТВА
Супрамолекулярная (надмолекулярная) химия – междисциплинарная область знания, синтезирующая достижения и терминологию целого ряда естественно-научных дисциплин (рис. 2.1).
Рис. 2.1. Основы супрамолекулярной химии
«Супрамолекулярная химия – это «химия за пределами молекулы», изучающая структуру и функции ассоциаций двух или более химических частиц, удерживаемых вместе межмолекулярными силами», − такое определение дал в 1989 году Ж.-М. Лен, ранее предложивший сам термин «супрамолекулярная химия». В настоящее время определений супрамолекулярной химии множество, поскольку исследователи пытаются дать наиболее точное научное определение.
В супрамолекулярной химии молекула играет роль, аналогичную той, которую атом выполняет в традиционной химии, то есть является как бы неделимой частицей в надмолекулярных процессах. Взаимодействуя между собой, эти молекулы хотя и претерпевают определенные изменения, но в таких пределах,
61
которые, как правило, оставляют за ними их химическую индивидуальность. На рис. 2.2 представлено сравнение диапазонов молекулярной и супрамолекулярной химии.
Рис. 2.2. Сравнение диапазонов молекулярной и супрамолекулярной химии
Межмолекулярные взаимодействия могут привести к образованию макромолекулярного ансамбля (надмолекулы), который состоит из нескольких десятков молекул, или (при хорошей пространственной комплементарности) к образованию клатратных соединений. Последние представляют собой молекулярные кристаллы, построенные из разного сорта молекул таким образом, что молекулы одного сорта строят кристаллический каркас (молекулы-«хозяева»), в полостях которого располагаются мо- лекулы-«гости». Клатраты относятся к так называемым гетеромолекулярным кристаллам (в отличие от гомомолекулярных кристаллов − молекулярных кристаллов, построенных из одного сорта молекул). Упаковка молекул разного сорта с образованием кристаллической клатратной фазы энергетически более выгодна, чем существование исходных веществ порознь в присущем им при данных условиях агрегатном состоянии.
Согласно Ж.-М. Лену, супрамолекулярную химию можно разбить на две широкие, частично налагающиеся друг на друга области:
62
–химию супермолекул – четко обозначенных олигомолекулярных частиц, возникающих в результате межмолекулярной ассоциации нескольких компонентов – рецептора и его субстрата («хозяина» и «гостя» – по другой терминологии) и строящихся по принципу молекулярного распознавания;
–химию молекулярных ансамблей – полимолекулярных систем, которые образуются в результате спонтанной ассоциации неопределенного числа компонентов с переходом в специфическую фазу, имеющую более или менее четко обозначенную микроскопическую организацию изависимые от ее природы характеристики (например, клатраты, мембраны, везикулы, мицеллы).
Основные функции супермолекул: молекулярное распознавание, превращение (катализ) и перенос. Функциональные супермолекулы наряду с организованными полимолекулярными ансамблями и фазами могут быть использованы для создания молекулярных и супрамолекулярных устройств.
2.1. Исторический экскурс
Супрамолекулярная химия – одна из самых молодых дисциплин, появление которой относят к 60–70 годам XX века. Однако истоки основных понятий супрамолекулярной химии можно найти в работах, выполненных значительно раньше (табл. 2.1).
|
|
Таблица 2 . 1 |
Некоторые этапы развития супрамолекулярной химии |
||
|
|
|
Год |
Автор |
Достижение |
1810 |
Г. Дэви |
Открытие гидрата хлора |
1823 |
М. Фарадей |
Формула гидрата хлора |
1893 |
А. Вернер |
Химия координационных соединений |
1894 |
Э. Фишер |
Концепция «ключ-замок» (молекулярное |
|
|
распознавание) |
1906 |
П. Эрлих |
Введение понятия «рецептор» (селективное |
|
|
связывание) |
1937 |
К. Вольф |
Введениетермина«супермолекула» дляопи- |
|
|
санияобразований, возникающихприобъеди- |
|
|
нениикоординационнонасыщенныхчастиц |
|
|
63 |
|
|
Окончание табл. 2 . 1 |
Год |
Автор |
Достижение |
1961 |
Н. Куртис |
Первое макроциклическое основание Шиф- |
|
|
фа из ацетона и этилендиамина |
1964 |
В. Буш и В.-Г. Егер |
Макроциклические основания Шиффа |
1967 |
Ч. Педерсен |
Синтез краун-эфиров |
1978 |
Ж.-М. Лен |
Введение понятия «супрамолекулярная хи- |
|
|
мия» и ее определения «химия молекуляр- |
|
|
ных ансамблей и межмолекулярных связей» |
1983 |
Д. Крам |
Синтез сферандов и кавитандов |
1987 |
Присуждение Нобелевскойпремии похимииД. Краму, Ж.-М. Лену |
|
|
иЧ. Педерсену заихработывобластисупрамолекулярнойхимии |
|
По мнению Ж.-М. Лена, фундамент супрамолекулярной химии заложили три понятия – селективное связывание (П. Эрлих, 1906), распознавание (Э. Фишер, 1894) и координация (А. Вер-
нер, 1893).
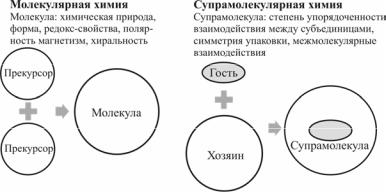
На рис. 2.3 представлены четыре фундаментальные системы супрамолекулярной химии, в трех из которых использованы реакции конденсации альдегидов и аминов с образованием иминов (основания Шиффа). Эти системы можно рассматривать как аналоги природных макроциклов (ионофоров, гемов, фталоцианинов и др.). Позднее к этим системам добавились криптанды, сферанды и кавитанды.
Соединения «хозяева» могут быть разделены на 2 главных класса в соответствии с топологической взаимосвязью между «хозяином» и «гостем» (рис. 2.4).
Кавитандами называют молекулы-«хозяева» с внутримолекулярными полостями. Способность полости связывать молеку- лу-«гостя» в этом случае – неотъемлемое свойство «хозяина». Она существует и в твердом состоянии, и в растворе. Клатрандами называют «хозяев» с межмолекулярными полостямизазорами между двумя или более молекулами «хозяев», существующими только в кристаллическом или твердом состоянии. Агрегат «хозяин»–«гость», образованный кавитандом, называют кавитатом, образованный клатрандом – клатратом. На рис. 2.5 приведены основные встречающиеся в литературе термины, описывающие взаимоотношения между «хозяином» и «гостем».
64
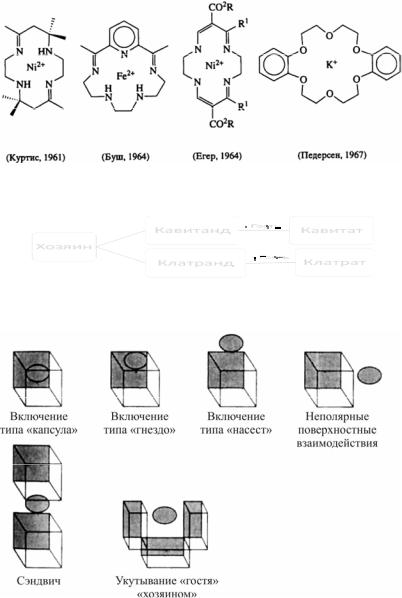
Рис. 2.3. Фундаментальные системы супрамолекулярной химии
Рис. 2.4. Основные классы соединений хозяин-гость
Рис. 2.5. Основные встречающиеся в литературе термины, описывающие взаимоотношения между «хозяином» и «гостем»
65
2.2. Природа супрамолекулярных взаимодействий
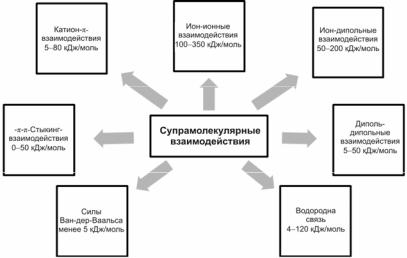
Супрамолекулярная химия имеет дело с нековалентными связывающими взаимодействиями, т.е. с разнообразными силами притяжения и отталкивания (рис. 2.6).
Рис. 2.6. Супрамолекулярные взаимодействия и их энергия связи
При анализе супрамолекулярной системы очень важно учитывать взаимовлияния этих взаимодействий и эффекты, относящиеся не только к хозяину и гостю, но и к их окружению (сольватация, кристаллическая решетка и т.д.).
Ион-ионные взаимодействия
По силе ионная связь (энергия связи 100–350 кДж/моль) сравнима с ковалентной. Типично ионное твердое тело – хлорид натрия – может быть представлено как супрамолекулярное соединение, в котором катион натрия организует вокруг себя шесть комплементарных донорных атомов. В растворе структура этого типа распадается из-за эффектов сольватации.
66
Более наглядным примером супрамолекулярных ионионных взаимодействий являются координационные соединения со сложными лигандами.
Ион-дипольные взаимодействия
Примером такого взаимодействия (энергия связи 50– 200 кДж/моль) может служить связывание иона с полярной молекулой с образованием комплекса. Притяжение полярных молекул к ионам гораздо сильнее, чем к другим полярным молекулам, особенно если ион многозарядный.
Ион-дипольные взаимодействия хорошо изучены в растворах полярных растворителей. Под действием электрического поля иона полярные молекулы растворителя ориентируются соответствующим образом, создавая вокруг каждого иона сольватную оболочку, в которую входит определенное число молекул растворителя, прочно связанных с ионом и участвующих в тепловом движении вместе с ним как единое целое. Энергия ион-дипольных взаимодействий тем больше, чем больше заряд и меньше радиус иона. Наличие вокруг иона сольватной оболочки и высокая диэлектрическая проницаемость растворителя препятствуют рекомбинации (объединению в молекулу) ионов.
Одна из самых полярных молекул – молекула воды, поэтому в водных растворах мы чаще всего имеем дело с супрамолекулярными структурами, называемыми гидратами, которые могут существовать не только в растворе, но и в виде кристаллов. У раствора при образовании гидратов появляются новые свойства, отсутствующие у компонентов. Например: CuSO4 и вода бесцветны, а их раствор (и кристаллогидрат) голубой; уксусная кислота и вода очень плохо проводят ток, а их раствор – гораздо лучше и т.д.
На рис. 2.7 приведены структурные формулы комплексов катиона натрия с молекулами воды и краун-эфиром, связанные ион-дипольным взаимодействием. В краун-эфирах при образовании комплексов эфирные атомы кислорода играют ту же роль, что и полярные молекулы воды.
67

Рис. 2.7. Na+ – комплексы с молекулами воды (а) и краун-эфиром (б)
Диполь-дипольные взаимодействия
Диполь-дипольные взаимодействия (энергия связи 5– 50 кДж/моль) могут возникать при ориентации одного диполя относительно другого. При этом притягиваются либо два полюса соседних молекул (I тип), либо два диполя (II тип). Такое поведение характерно, в частности, для органических карбонильных соединений (рис. 2.8).
Рис. 2.8. Диполь-дипольные взаимодействия в карбонилах
Водородная связь
Водородную связь относят к числу слабых химических взаимодействий (энергия связи 4–120 кДж/моль), тем не менее она играет исключительную роль в стабилизации конденсированных состояний многих простых молекулярных систем, например воды, и в стабилизации биополимеров (нуклеиновых кислот, белков). Водородную связь можно рассматривать как особый вид диполь-дипольного взаимодействия, в котором атом водорода, присоединенный к электроотрицательному атому (или к группе атомов) притягивается к диполю соседней молекулы или функциональной группы. Благодаря такой направленной и относительно сильной природе водородная связь считается «ключевым взаимодействием в супрамолекулярной химии».
68
Водородные связи позволяют полимерным цепям соединяться в специфические трехмерные структуры, приобретающие при этом функциональную биологическую активность. Эти структуры, с одной стороны, достаточно прочные (за счет образования большого числа водородных связей), а с другой − достаточно чутко реагирующие на изменение внешних условий (например, приближение той или иной молекулы) именно из-за того, что эти взаимодействия являются слабыми. Разрыв таких связей лишает белки или нуклеиновые кислоты их биологических функций.
Современные расчеты показывают, что суммарный заряд на атоме водорода, участвующем в образовании водородной связи, практически не меняется по сравнению с зарядом в мономерной молекуле, что говорит о том, какую заметную роль в образовании водородной связи должны играть поляризация, перераспределение электронного заряда в отдельных областях пространства. Таким образом, водородная связь по своему происхождению не представляет собой нечто, отличающееся от того, что характерно для химических связей вообще. Ее определяют главным образом поляризация электронного распределения в мономерных звеньях (в общем случае в молекулах, образующих такую связь) и отличная от мономерных звеньев динамика колебательного движения атомов в водородно-связанном фрагменте.
Расстояние между соседними атомами, участвующими в образовании водородной связи, значительно меньше суммы их вандерваальсовых радиусов. Так, в воде расстояние между атомами кислорода в системе О-Н_О составляет 0,276 нм. Если принять, что длина ковалентной связи О-Н равна 0,1 нм, то длина связи Н_О составит 0,176 нм, то есть она значительно (примерно на 70 %) длиннее ковалентной связи между этими атомами. Тем не менее связь Н_О оказывается значительно короче суммы вандерваальсовых радиусов, составляющих для водорода и кислорода соответственно 0,12 и 0,14 нм. Последнее обстоятельство является одним из критериев, указывающих на образование между молекулами водородных связей.
69
Водородная связь увеличивает длину связи Х–Н, что приводит к смещению соответствующей полосы валентных колебаний в ИК-спектре в сторону более низких частот. Метод ИКспектроскопии является главным методом изучения водородной связи. Протоны, участвующие в водородной связи, характеризуются более низкой электронной плотностью, поэтому они деэкранируются, что приводит к существенному смещению соответствующих резонансных сигналов в спектрах ЯМР.
Водородные связи широко представлены в органической химии. Все органические соединения за самым редким исключением содержат водород, то есть являются кислотами Бренстеда, а наиболее часто входящие в их состав элементы-органогены (O, N, S, галогены) содержат неподеленные пары электронов и могут выступать в качестве основных центров. Учитывая отмеченное, можно сказать, что большинство органических соединений потенциально способно к образованию водородных связей. По структурной формуле (природа взаимодействующих групп и их взаимное расположение) можно предсказать силу водородных связей и их характер (внутриили межмолекулярные). При оценке взаимного влияния атомов в молекулах обязательно учитываются возможность образования водородных связей и их последующее влияние на скорость, механизм и направление реакций. Оценить влияние среды (растворителя) на ход химического процесса часто становится возможным лишь с учетом образования водородных связей.
Катион-π-взаимодействия
Взаимодействия катионов щелочных и щелочноземельных металлов с двойной связью С=С носят название «катион-π- взаимодействия» (энергия связи 5–80 кДж/моль). Они играют очень важную роль в биологических системах.
π-π-Стэкинг-взаимодействия
Это слабое электростатическое взаимодействие (энергия связи 0–50 кДж/моль) часто наблюдается между ароматическими кольцами, если одно из них богато электронами, а второе – испытывает их недостаток. На рис. 2.9 приведены два основных
70