

Российский химико-технологический университет им. Д.И. Менделеева Кафедра квантовой химии
Компьютерное моделирование процессов нанотехнологий.
Лекция 6. Полуэмпирическая квантовая химия.
Цирельсон В.Г. , Бобров М.Ф. «Квантовая химия молекул».
Москва 2007 г.
1

Методы вычислительной химия наноразмерных систем
неэмпирическая |
молекулярная |
|
квантовая химия |
||
механика |
||
|
квантовая |
молекулярная динамика |
|
статистическая |
||
и метод Монте-Карло |
||
механика |
||
|
2

Полуэмпирическая квантовая химия Идея: 1) вместо точного оператора Фока - приближенный, элементы
которого получают из эмпирических данных; параметры одно- и двухэлектронных интегралов подбирают для каждого атома или для пар атомов.
2) многоэлектронная волновая функция – один детерминант, базисные функции i – симметричные ортогональные комбинации ОСТ { j }, которые получаются из исходных ОСТ с помощью преобразования Левдина = S-1/2.
Расчет МО проводится обычным итерационным путем.
Важно: параметры справедливы лишь в пределах узкого класса соединений, поэтому придавать им физический смысл не следует.
Основные приближения полуэмпирических методов 1) Рассматриваются только валентные электроны: электроны
атомных остовов включают в функции, описывающие энергию отталкивания остов-остов. Поляризацией остовов пренебрегают.
2) Базис считают минимальный, полагая, что базисные функции
3
образуют набор ортогональных АО.

3) Для двухэлектронных интегралов вводят приближение Нулевого
Дифференциального Перекрывания (НДП)
( r ) ( r )dr =0.
Считают, что из-за экспоненциального убывания АО двухэлектронными кулоновскими и обменными интегралами, содержащими произведения различных атомных орбиталей, зависящих от одного аргумента, можно пренебречь:
( ) = ( ) = ( )
В приближении НДП, принимаемом для всех пар АО, уравнения Рутана имеют
вид: |
ci (F |
Ei δ ) 0 |
|||
|
|||||
|
|
|
|
|
|
|
Элементы матрицы Фока: |
|
|
|
|
|
F h |
1 P (μμ|νν) P (μμ|νν) |
|||
|
|
2 |
|
|
|
|
|
|
|
|
μ ν |
|
F |
h |
|
|
1 P (μμ|νν) |
|
|
|
|
|
2 |
4

4) Результат расчета не должен зависеть от выбора декартовой системы
координат, в которой определяются ориентации p-, d- и т.д. АО. НДП нарушает это требование. Например, двухцентровый кулоновский интеграл (px py s2), где px и py - АО одного и того же атома в приближении НДП
должен быть равен нулю. Повернем теперь декартову систему координат вокруг
оси z на угол : тогда |
p x = |
px cos + py sin |
p y = px sin - py |
cos |
и |
(p x p y s2) = - (p 2x s2) cos sin + (p 2y s2) cos sin + (px py s2)( cos2 - sin2 )= [(p 2y s2) - (p 2x s2) ] cos sin
(s-АО при повороте не меняется, px и py АО ортогональны). (2.49) равно нулю только при инвариантен относительно поворота системы
Иллюстрация происхождения неинвариантности двухцентрового кулоновского интеграла по отношению к повороту осей координат
5

Вращательная инвариантность нарушается каждый раз, когда двухэлектрон- ные интегралы включают перекрывание двух АО одного и того же атома.
Поэтому в таких случаях вводится еще одно приближение: считают, что двухэлектронные интегралы ( ) зависят только от природы атомов, на которых центрированы орбитали и , и не зависят от конкретного вида
орбиталей.
Это соответствует сферическому усреднению распределения валентных электронов на АО различных атомов молекулы при расчете взаимодействия и обеспечивает инвариантность решения относительно поворота систем координат.
Для усредненного интеграла ( ) используется обозначение AB ,
где А и В обозначают атомы, на которых центрированы АО интегралы и ;
вычисляется он с s-АО соответствующих атомов
AB = (s2A s2B)
Для одноэлектронных интегралов также вводятся различные приближения,
которые рассмотрим далее применительно к конкретным методам. |
6 |
|

Приближение НДП для двухэлектронных интегралов Двухэлектронные интегралы в приближении НДП принимают равными
нулю, если они включают перекрывание пар АО разных атомов или пары разных АО одного и того же атома. Однако для р-АО вращательная инвариантность нарушается в любом случае. Поэтому вводится еще одно приближение: считают, что двухэлектронные интегралы ( ) зависят только от природы атомов, на которых центрированы орбитали и , и не зависят от конкретного вида
орбиталей. Это соответствует сферическому усреднению распределения валентных электронов на АО различных атомов молекулы при расчете взаимодействия и обеспечивает инвариантность решения относительно поворота систем координат.
Для усредненного интеграла ( ) используется обозначение AB , где А и В обозначают атомы, на которых центрированы АО интегралы и .
Интеграл AB вычисляется с помощью s-АО соответствующих атомов
AB = (s2A s2B)
или по специальным эмпирическим формулам.
7

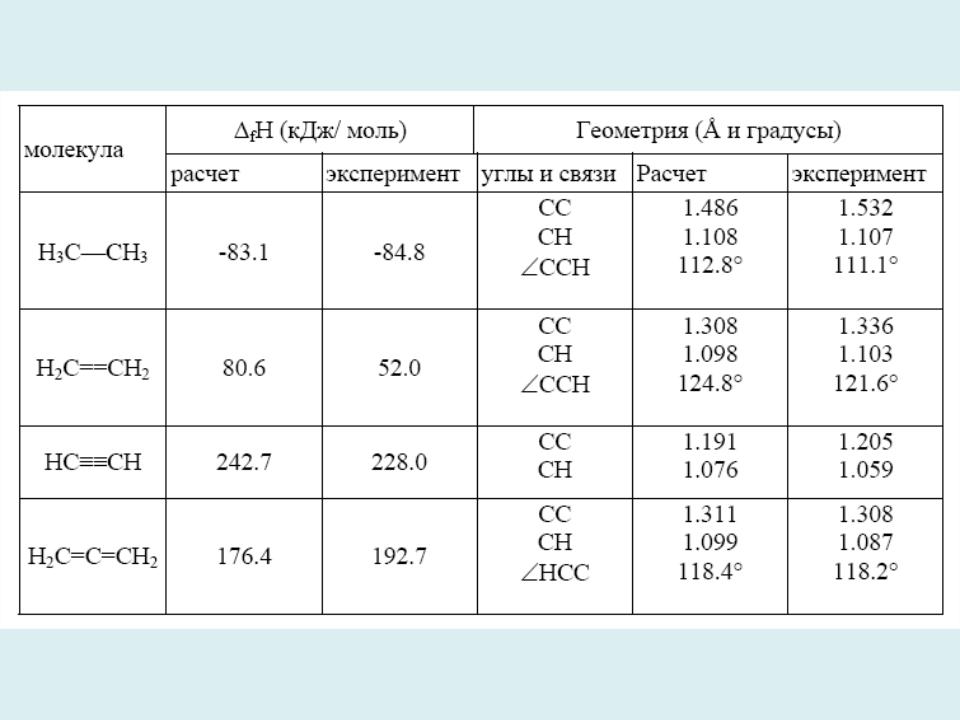
Теплоты образования и геометрические характеристики некоторых молекул (MINDO/3)
9

Длины связей (A), валентные углы (град.) молекул, рассчитанные по MINDO/3, неэмпирически в базисе STO-3G и полученные экспериментально
10