2.Репарация днк. Этапы репарации. Дефекты репарации.
Репарация необходима для сохранения генетического материала на протяжении все жизни.
ДНК постоянно подвергается разнообразным изменениям, вызванным действием различных веществ внешней и внутренней среды, радиацией и УФ. И под данным действием в структуре ДНК происходит:
· Дезаминирование оснований , при котором цитозин превращается в урацил, аденин-вгипоксантин, а гуанин- в ксантин.
· Депуринизация или депиримидинизация, результатом котрой является появление в ДНК остатков дезоксирибозы, лишенных основания
· Образование пирмидиновых димеров под действием УФ
· Разрыв нуклеотидных цепей
· Появление ковалентных сшивок между цепями и гистонами
· Появление ошибок репликации
· Образование продуктов алкилирования ДНК
ДНК-N-гликозилазами обнаруживаются и удаляются поврежденные основания ДНК. Ферменты гидролитически расщепляют N-гликозидную связь между поврежденным основанием и остатком дезоксирибозы. Участи ДНК, лишенные азотистого основания, получили название АП-сайтов. Они могут также возникать в результате самопроизвольногогидролитического отщепления пуриновых или пиримидиновых оснований. Далее ход репарации идет по одному из путей
· Либо фермент ДНК-инсертаза может присоединять к дезоксирибозе основание в соответствии с правилом комплиментарности
· Либо эндонуклеаза определяет место повреждения и гидролизует 3,5 – фосфодиэфиную связь
Эндонуклеаза находит место разрыва цепи и удаляет поврежденный участок. ДНК-полимраза в(бета) присоединяется к 3-концу образовавшейся «бреши» и достраивает недостающий участок цепи. Матрицей служит неповрежденная цепь ДНК.
ДНК-лигаза соединяет неповрежденный вновь синтезированный участки цепи ДНК.
*если одновременно повреждаются комплементарная пара нуклеотидов, репарация в гаплоидных клетках невозможна, а в диплоидных может идти за счет присутствия идентичного гена в гомологичной хромосоме
Снижение активности ферментов репарации приводит к накоплению поврежденй в ДНК
При недостаточность ферментов УФ-эндонуклеазы и полимеразы-1, которые отвечают за восстановление ДНК после ее повреждения УФ-лучами, развивается пигментная ксеродерма(поражение кожи и глаз)
При нарушении экцизионной репарации(удаляет неправильно спаренные или повреждённые основания из ДНК с последующим синтезом новой последовательности по неповреждённой цепи) могут развиваться синдром Коккёйна(карликовость, атрофия зрительного нерва, раннее старение), трихотиодистрофия.
При нарушении рекомбинационной репарации( для восстановления одной хромосомы используется материал гомолога) может развиваться синдром Блума(повышенная чувствительность к УФ, частая заболеваемость лейкемией).
3) Понятие о рекомбинантных ДНК.
Задача.
Одним из первых синтетических бактериостатических препаратов был сульфаниламид (стрептоцид). На чём основано бактериостатическое действие данного вещества?
Ответ:
Сульфаниламидны, являясь структурными аналогами параминобензойной кислоты (ПАБК), действуют как антиметаболиты, приводя к синтезу аналога фолиевой кислоты, не обладающего её функциями, что делает размножение бактерий невозможным. Основой для синтеза фолиевой кислоты служит парааминобензойная кислота (ПАБК), которую бактерии получают из среды.
Билет б/н (5)
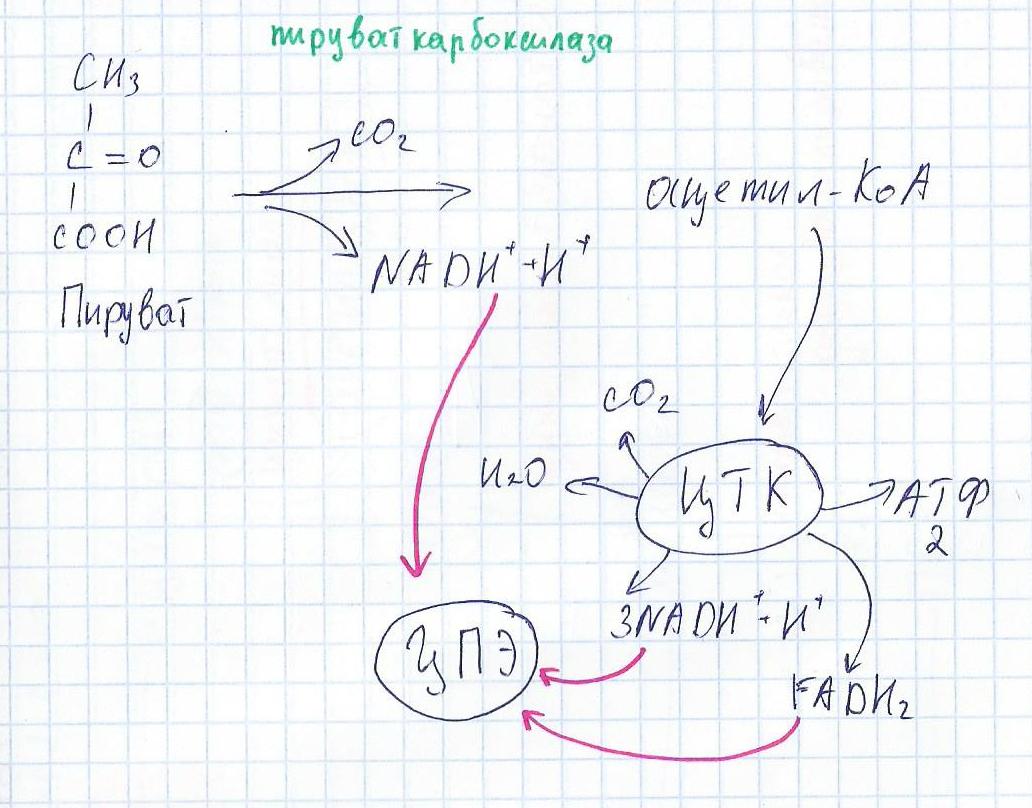
1) Окислительное декарбоксилирование пировиноградной кислоты: схема процесса, связь с синтезом АТФ. Строение пируватдегидрогеназного комплекса: ферменты, коферменты, регуляция процесса. Если в процессе катаболизма белков, жиров, углеводов образуется ПВК, для дальнейшего окисления, необходим переход её в ацетил КоА. Этот процесс называется окислительным декарбоксилированием пировиноградной кислоты. В нём заключено два вида реакций: окисление и образование СО2 путём разрушения карбоксильной группы. Окислительное декарбоксилирование пирувата осуществляется при участии пируватдегидрогеназного комплекса.
Состав полиферментного пируватдегидрогеназного комплекса Пируватдегидрогеназный комплекс (ПДК) молекулярной массой 6*106 дальтон, включает в себя три вида ферментов (Е1-Е3) и пять видов коферментов. 2 кофермента НАД и HS-КоА находятся в свободном состоянии и входят в состав комплекса только в момент реакции. Общий вид реакции окислительного декарбоксилирования пирувата:
Ферменты пируватдегидрогеназного комплекса:
Е1 – пируватдегидрогеназа (пируватдекарбоксилаза)
Е2 – дигидролипоилацетилтрансфераза
(трансацетилаза)
Е3 – дигидролипоилдегидрогеназа
Коферменты пируватдегидрогеназного комплекса:
1. Тиаминдифосфат (ТДФ, ТПФ), содержащий витамин В1,
кофактор пируватдегидрогеназы.
2. Липоевая кислота, кофактор трансацетилазы.
3. Кофермент ФАД, содержащий витамин В2, кофактор
дегидрогеназы дигидролипоевой кислоты.
4. Кофермент НАД, содержащий витамин РР.
5. Кофермент НS-КоА, содержащий аденин, рибозу, два
остатка фосфорной кислоты, пантотеновую кислоту
(витамин В3).
Окислительное декарбоксилирование ПВК протекает в несколько стадий, в процессе которых двухуглеродный фрагмент, образующийся из ПВК, переносится на липоевую кислоту, а затем на HS-КоА.
Биологическая роль окислительного декарбоксилирования пирувата заключается в том, что оно является важным этапом катаболизма, позволяющим включаться в цикл Кребса тем веществам, при распаде которых образуется ПВК. Образовавшаяся молекула НАДН2 окисляется в длинной дыхательной цепи с образованием 3-х молекул АТФ.
Окислительное декарбоксилирование пирувата протекает внутри митохондрий. Регуляция пируватдегидрогеназного комплекса осуществляется путём фосфолирирования, дефосфолирирования пируватдегидрогеназы Активаторами ПДК служат АДФ и НАД окисленный. Ингибиторами этого комплекса являются АТФ и НАДН2..
В состав ПДК входит пять витаминов.
По механизму «обратной связи» работу пируватдегидрогеназного комплекса ингибируют конечные продукты окислительного декарбоксилирования - ацетил-KоА, НАДН + Н+, а также АТФ. Увеличивает активность комплекса пировиноградная кислота. Также имеется регуляция со стороны гормонов: инсулин увеличивает активность комплекса, глюкагон - снижает. Первую реакцию катализирует Е1, субстратами являются ПВК и дегидролипоевая кислота, являющаяся простетической группой Е2. От ПВК отщепляется карбоксильная группа и образуется СО2, а ацетильный остаток соединяется с атомомсеры липоевой кислоты в составе ацетилтрансферазы. Получается ацетиллипоат-Е2.
Во второй реакцииацетилтрансфераза (Е2) катализирует перенос ацетильного остатка, соединенного с его собственной простетической группой, на коэнзим А. Продукты этой реакции - дигидролипоевая кислота в составе Е2 и ацетил-КоА.
В третьей реакциипроисходит дегидрирование дигидролипоевой кислоты в составе ацетилтрансферазы при воздействии фермента Е3 (дегидрогеназа дигидролипоевой кислоты), содержащего ФАД. ФАД передает водород на НАД. Образуются НАДН, Н+ и дегидролипоевая кислота в составе Е2. Последний фермент снова вступает в окислительное декарбоксилирование ПВК.
2) Молекулярные механизмы малигнизации клеток. Канцерогенез - это процесс развития опухолей любого типа. Последняя стадия опухолевого роста, с видимыми проявлениями , манифестация получил название малигнизации ( озлакочествление). Общие признаки малигнизации:
1. Клетка приобретает способность к бесконтрольному , безудержному размножению, делению
2. Гиперплазия параллельно с бесконтрольным делением клеток, наблюдается нарушение дифференцировки, остается незрелой, молодой ( это свойство называется анаплазией).
3. Автономность ( независимый от организма), от контролирующей, регулирующей процессы жизнедеятельности стимулов. Чем быстрее растет опухоль, тем как правило менее дифференцированны клетки и больше выражена автономность опухоли.
4. Доброкачественная опухоль характеризуется нарушением пролиферации, нет нарушения дифференцировки, при росте доброкачественной опухоли клетки просто увеличиваются в количестве, раздвигая или сдавливая окружающие ткани. А для злокачественных опухолей характерен так называемый инфильтративный рост, опухолевые клетки прорастают ( как клетки рака) разрушая окружающие ткани.
5. Способность к метастазированию. Метастазы - это клетки которые могут гематогенным, лимфогенным путем разноситься по всему организму и образовывать очаги опухолевого процесса. Метастазы - это признак злокачественной опухоли.
6. Опухолевая ткань оказывает на организм в целом негативное влияние : интоксикация, вызванная продуктами метаболизма опухоли, распада опухоли. Кроме того опухоль лишает организм необходимых питательных веществ, энергетических субстратов, пластических компонентов.
Атипизм опухолевых клеток характеризуется как возврат к прошлому то есть переходом на более простые пути метаболизма . Существует множество признаков, отличающих нормальные клетки от опухолевых:
1. Морфологический атипизм. Главным является изменение клеточной мембраны:
У опухолевых клеток уменьшается площадь поверхности соприкосновения, уменьшается количество нексусов - контактов, обеспечивающих адгезивность клеточных мембран, меняется состав мембранных гликопротеидов - укорачиваются углеводные цепи. В клетке начинают синтезироваться , несвойственные зрелым клеткам эмбриональные белки, повышается количество фосфотирозинов. Все это приводит и к нарушению свойств контактного торможения, повышается лабильность, текучесть мембраны. В норме клетки, вступая в контакт друг с другом прекращают деление ( имеет место саморегуляция процесса деления). В опухолевых клетках отсутствие контактного торможения приводит к безудержной пролиферации.
2.Биохимический атипизм. Атипизм энергетического обмена проявляется в преобладании гликолиза - более древнего пути метаболизма. В опухолевых клетках наблюдается интенсивный анаэробный гликолиз Опухоль активно поглощает питательные вещества. Наблюдается феномен субстратных ловушек, который заключается в повышении сродства фермента к субстрату ( глюкозе), в опухолевых клетках в 1000 раз повышается активность гексокиназ. Клетки опухоли являются также ловушкой для белка что также приводит к кахексии.
Преобладание гликолиза приводит к повышению концентрации молочной кислоты в клетках опухоли, характерен ацидоз, приводящий к нарушению жизнедеятельности самой клетки ( зона некроза расположена обычно в центре опухоли).
Возрастает синтез рибонуклеотид – редуктазы, снижается катаболизм пиримидиновых и пуриновых нуклеотидов, увеличивается синтез ДНК и РНК, увелчивается количество фетальных форм ферментов
3.Атипизм регуляции роста и дифференцировки опухолевых клеток. Процессы роста , дифференцировки деления в норме находятся под контролем центральной эндокринной регуляции, которая осуществляется соматотропным гормоном, гормонами щитовидной железы, инсулином. Кроме этих общих факторов , в каждой ткани существуют свои факторы роста и дифференцировки ( фактор роста эпидермиса, тромбоцитарный фактор, интерлейкин). Индукция роста и дифференцировки начинается с взаимодействия фактора роста с рецептором фактора роста на клеточной мембране ( в опухолевой клетке этот этап может быть нарушен). На следующем этапе образуются вторичные посредники - циклический аденозин и гуанозинмонофосфат, причем для нормального роста и дифференцировки характерно преобладание циклического аденозинмонофосфата ( цАМФ). Образование циклического гуанозинмонофосфата сочетается с усилением пролиферации. В опухолевых клетках это типичный признак.
Регуляция роста и дифференцировки опухолевой клетки связана также с кальций-зависимой протеинкиназой. В норме кальций-зависимая протеинкиназа выполняет функцию модулятора, она выполняет роль индуктора пролиферации, она стимулирует образование фосфотирозина и усиливает
3) Антидиуретический гормон (вазопрессин): химическая природа, механизм действия, органы-мишени, биологические эффекты. Несахарный диабет. Антидиуретический гормон:
Строение:
Представляет собой пептид, включающий 9 аминокислот, с периодом полураспада 2-4 минуты.
Синтез:
Осуществляется в супраоптическом и паравентрикулярном ядрах гипоталамуса. Отсюда в точку секреции (заднюю долю гипофиза) вазопрессин отправляется в виде прогормона, состоящего из двух частей – собственно АДГ и нейрофизина. В ходе транспортировки происходит процессинг – гидролиз проАДГ на зрелый гормон и белок нейрофизин.
Регуляция синтеза и секреции:
Уменьшают: этанол, глюкокортикоиды.
Активируют:
• возбуждение осморецепторов в гипоталамусе и в портальной вене печени из-за повышения осмолярности плазмы при обезвоживании, почечной или печеночной недостаточности, накоплении осмотически активных веществ (глюкоза),
• активация барорецепторов сердца и каротидного синуса при снижении объема крови в сосудистом русле (кровопотери, обезвоживание),
• эмоциональный и физический стресс,
• никотин, ангиотензин II, интерлейкин 6, морфин, ацетилхолин.
Механизм действия:
Зависит от рецепторов:
1. Кальций-фосфолипидный механизм, сопряжен:
• с V1-рецепторами гладких мышц артериол, печени, тромбоцитов,
• с V3-рецепторами аденогипофиза и структур головного мозга.
2. Аденилатциклазный механизм – с V2-рецепторами почечных канальцев.
Мишени и эффекты:
Почки:
Увеличивает реабсорбцию воды в эпителиоцитах дистальных канальцев и собирательных трубочек, благодаря "выставлению" на мембрану транспортных белков для воды – аквапоринов:
• через аденилатциклазный механизм вызывает фосфорилирование молекул аквапоринов (только тип 2, AQP2), их взаимодействие с белками микротубул и путем экзоцитоза встраивание аквапоринов в апикальную мембрану,
• по тому же механизму стимулирует синтез аквапоринов de novo.
Сосудистая система:
Поддерживает стабильное давление крови, стимулируя тонус сосудов:
• повышает тонус гладких мышц сосудов кожи, скелетных мышц и миокарда (в меньшей степени),
• повышает чувствительность механорецепторов в каротидных синусах к изменениям артериального давления,
Иные эффекты:
Метаболические эффекты:
Избыточное количество вазопрессина в крови:
• у голодных животных в печени активирует гликогенолиз, что вызывает выход глюкозы в кровь,
• у сытых животных в печени стимулирует гликолиз, который здесь является началом синтеза ТАГ и холестерола,
• усиливает секрецию глюкагона,
• понижает липолитический эффект катехоламинов в жировой ткани,
• усиливает секрецию АКТГ и, следовательно, синтез глюкокортикоидов.
В целом эффект вазопрессина на гормональный и метаболический статус организма сводится к гипергликемии и накоплению липидов.
Головной мозг:
• участвует в механизмах памяти и поведенческих аспектах стресса,
• через V3-рецепторы стимулирует в кортикотрофах секрецию АКТГи пролактина,
• повышает болевой порог чувствительности,
• повышение концентрации вазопрессина и дисбаланс вазопрессин/окситоцин отмечается при депрессии, тревоге, шизофрении, аутизме, расстройствах личности. В эксперименте вазопрессин вызывает у крыс агрессивное поведение и тревожность.
Костная ткань:
• Поддерживает обновление структур и минерализацию кости, усиливая активность как остеобластов, так и остеокластов.
Сосудистая система:
Влияет на гемостаз, в целом повышая вязкость крови:
• в эндотелии вызывает образование фактора Виллебранда, антигемофильного глобулина А (фактора свертывания VIII) и тканевого активатора плазминогена (t-PA),
• в печени также повышает синтез VIII фактора свертывания,
• усиливает агрегацию и дегрануляцию тромбоцитов.
Гипофункция проявляется в виде несахарного диабета, частота примерно 0,5% всех эндокринных заболеваний. Проявляется большим объемом мочи до 8 л/сутки, жаждой и полидипсией, сухостью кожи и слизистых, вялостью, раздражительностью.
Существуют разные причины гипофункции:
1. Первичный несахарный – дефицит АДГ при нарушении синтеза или повреждениях гипоталамо-гипофизарного тракта (переломы, инфекции, опухоли);
2. Нефрогенный несахарный диабет:
• наследственный – нарушение рецепции АДГ в канальцах почек,
• приобретенный – заболевания почек, повреждение канальцев солями лития при лечении больных психозами.
3. Гестагенный (при беременности) – повышенный распад вазопрессина аргинин-аминопептидазой плаценты.
4. Функциональный – временное (у детей до года) повышение активности фосфодиэстеразы в почках, приводящее к нарушению действия вазопрессина.
Билет 41
1) Трансляция: схема процесса, регуляция. Трансляция – это процесс синтеза белка из АК на матрице мРНК, осуществляемый рибосомой.
А)инициация начинается с присоединения к мРНК в области «кэпа»
малой субъединицы рибосомы 40S
факторов инициации,инициирующей Мет-тРНК
ГТФ
Когда в результате движения этого комплекса по мРНК антикодон Мет-тРНК свяжется с инициирующим кодоном AUG,комплекс останавливается.
Происходит присоединение 60S- субъединицы рибосомы, сопровождающееся гидролизом ГТФ и оотделением факторов инициации.
Формируется 80S-рибосома с 2 активными центрами
Р (пептидильным) центром, в котором находится Мет-тРНК
А(аминоацильным)центром, в область которого поступает первый смысловой кодон мРНК
Б)элонгация состоит из следующих стадий
Связывание аа-тРНК в А-центре. В свободный А-центр присоединяется первая аа-тРНК, у которой антикодон косплементарен кодону мРНК, находящемуся в области этого центра. Эта фаза процесса требует затраты энергии ГТФ и участия факторов элонгации EF1.
Образование пептидной связи. Происходит пептидилтрансферазная реакция, в ходе которой метионин от Мет-тРНК, входящей в Р-центр, переносится в а-аминогруппу АК (валина), находящейся в А-центре в составе аа-тРНК с образованием дипептидил тРНК
Транслокация. – перемещение рибосомы по мРНК. В ходе этой стадии рибосома сдвигается на один кодон в направлении от 5 к 3-концу мРНК за счет энергии ГТФ и при участии фактора элонгации EF2. В результате дипептидил-тРНК из А-центра попадает в Р-центр, а в А-центре оказывается следующий кодон.
Свободная тРНК теряет связь с Р-центром и покидает рибосому. Далее процесс продолжается по описанной схеме.
В)терминацияпроисходит псле включения в А-центр одного из стоп кодонов:UAG, UGA,UAA.
Белковые факторы терминации RF1, RF3, взаимодействуя с этими кодонами, при участии пептидил-ТФ обеспечивают гидролитическое отщепление синтезированного центра и диссоциацию рибосомы на субъединицы с затратой энергии молекулы ГТФ.
Одновременно несколько рибосом могут участвовать в трансляции одной мРНК. Каждая рибосома занимает участок, равный примерно 80 нуклеотидам мРНК. Т.О.рибосомы располагаются на мРНК с интервалом около 100 нуклеотидов, образуя комплекс-полисома
Функционально активные белки образуются в результате посттрансляционных модификаций ППЦ, синтезированных на рибосомах. Они включают:
Частичный протеолиз
Фолдинг, или формирование пространсвенной структуры, в котором принимают участие белки-шапероны, обеспечивающие образование функционально активной конформации ППЦ
Модификации АК:карбоксилирование, фосфорилирование, йодирование, гидроксилирование.
Образование дисульфидных связей между остатками цистеина, участвующими в формировании трехмерной структуры белка
Присоединение простетических групп
Образование олигомерных структур, которое также осуществляется при участии шапернов
2) Химическая модификация липидов и белков ЛПНП и рецепторов ЛПНП. Молекулярные механизмы развития атеросклероза. Коэффициент атерогенности. 3) Нарушение обмена АК
Билет 4 1.Регуляция ферментов 2.Особенности метаболизма глю в НТ 3.Механизм действия наркотиков ДОФА Задача ответ кетоацидоз из-за сахарного диабета
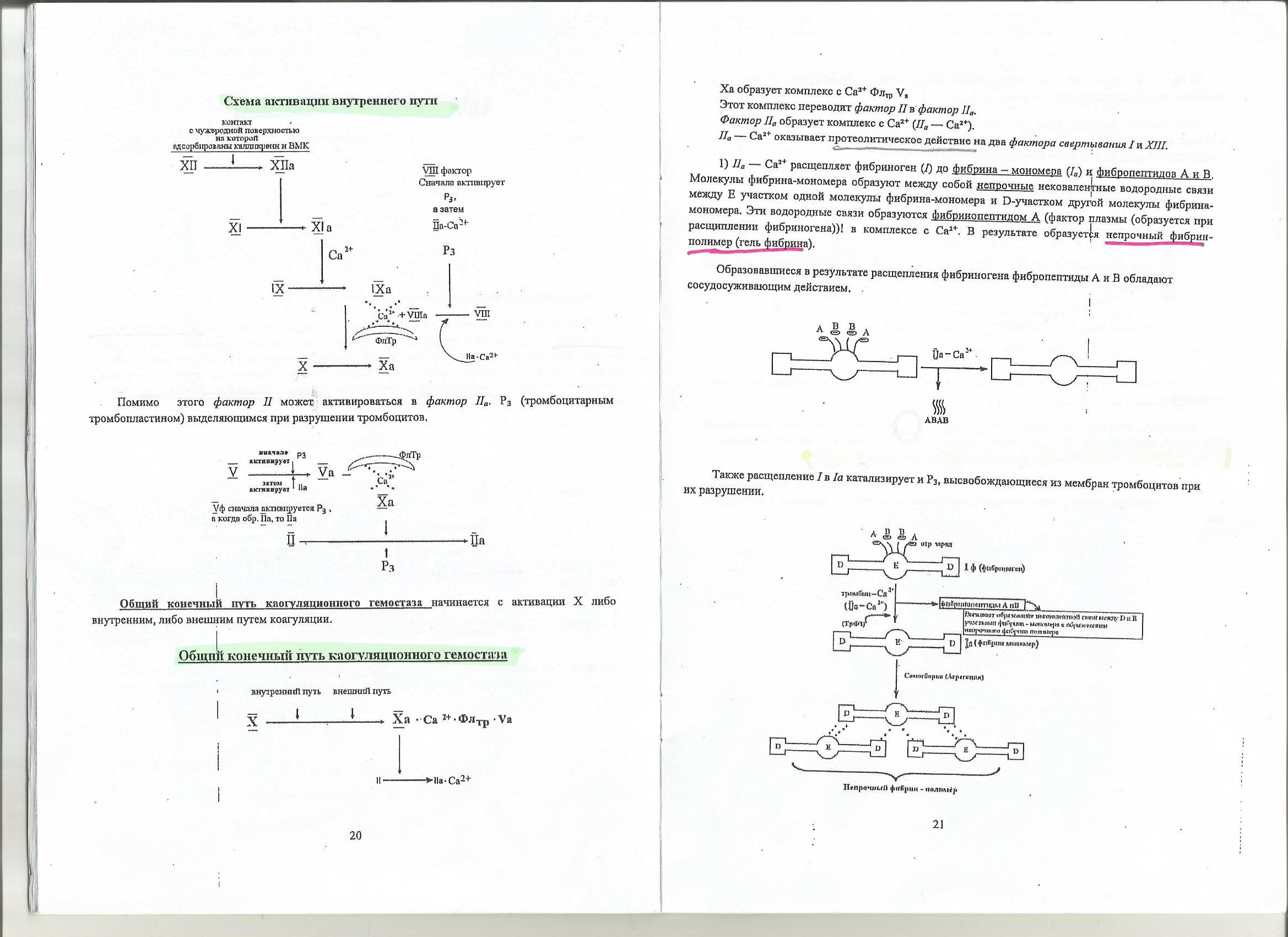
Билет 12 1.Гликолиз Гликолиз – это специфический путь катаболизма глюкозы, в результате которого происходит расщепление глюкозы с образованием
2 молекул пирувата - аэробный гликолиз
2 молекул лактата - анаэробный гликолиз
Все этапы гликолитического пути окисления глюкозы происходят в цитозоле.
Превращение глюкозы в 2 молекулы глицеральдегид-3-фосфата. Происходит потребление 2 АТФ
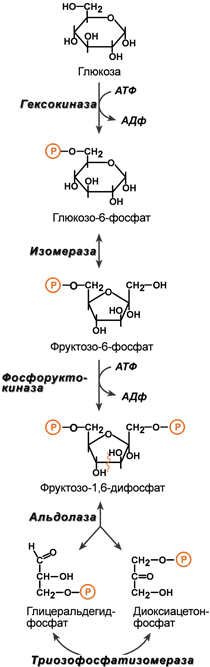
Превращение глицеральдегида в пируват или лактат.Происходит образование 2 АТФ, и НАДФН+Н+
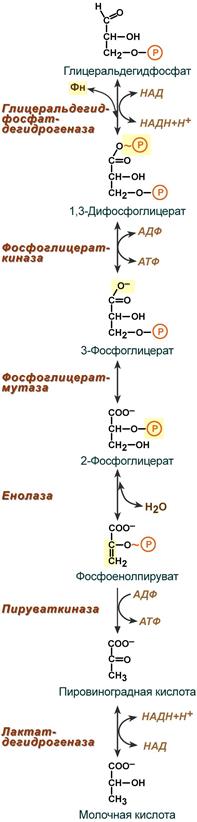
В результате 6 реакции образуется НАДФН+Н+. Передача водорода на ЦПЭ в митохондриях происходит за счет челночных систем:
Глицерофосфатная челночная система– водород передается на FAD-зависимую дегидрогеназу, Р/О=2
В цитозоле метаболиты
гликолиза – диоксиацетонфосфат и НАДН
образуют глицерол-3-фосфат,
поступающий в митохондрии. Там он
окисляется с образованием ФАДН2.
Далее ФАДН2 направляется
в
дыхательную цепь
и используется для получения энергии
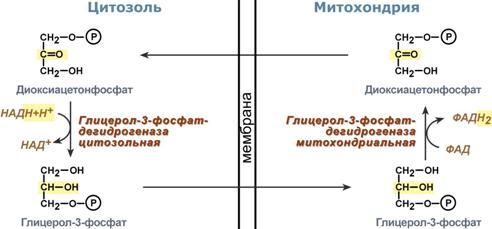
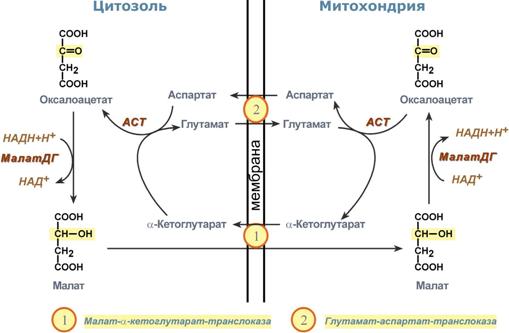
Малат-аспартатная челночная система – водород поступает в ЦПЭ через митохондриальный NAD и Р/О=3
В
цитоплазме NADH восстанавливает
оксалоацетат в малат, который при
участии переносчика проходит в
митохондрии, где окисляется в оксалоацетат
NAD-зависимой малатдегидрогеназой.
Восстановленный в ходе этой реакции
NAD отдаёт водород в митохондриальную
ЭТЦ. Однако образованный из малата
оксалоацетат выйти самостоятельно из
митохондрий в цитозоль не может, так
как мембрана митохондрий для него
непроницаема. Поэтому оксалоацетат
превращается в аспартат, который и
транспортируется в цитозоль, где снова
превращается в оксалоацетат.

Далее пируват окисляется до ацетил-КоА и поступает в ЦТК
Особенность метаболизма углеводов в нервной ткани.Глюкоза-основной субстрат для получения энергии в клетках нервной системы. Собственные запасы глюкозы в мозговой ткани очень малы, по сравнению с высокой активностью окисления. Поступает в нервную ткань путем облегченной диффузии.
85-90% глюкозы окисляется до воды и СО2, 5% расходуется в реакциях гликолиза с образованием молочной кислоты.
Основной метаболический путь - аэробный гликолиз. Поэтому при недостатке кислорода могут возникать нарушения деятельности мозга. Использование глюкозы зависит от активности гексокиназы(на 1 изоформу приходится 90% активности, на 2 – 10%). Несмотря на то что, ГМ – инсулиннезависимая ткань, при СД он испытывает некоорый недостаток(из-за того, что инсулин влияет на активность гексокиназы).
В глиальных клетках работает ЛДГ5 (находится в цитозоле) ПВК лактат(в сторону лактата)
В нейронах работает ЛДГ1 (находится в мх) ПВК лактат (в сторону ПВК)
Особенности энергетического обмена в нервной ткани.
ГМ составляет 2% от массы тела, но потребляет 20-25% кислорода. 3% кислорода от этого количества потребляет ПНС. 67% расходуемого кислорода приходится на нейроны. 30% - на глиальные клетки.
Активность фермента Mg2+-зависимой K+Na+-АТФазы выше в ГМ, чем в других тканях. Высокая потребность нейронов в энергии объясняется необходимостью синтеза нейромедиаторов.
В условиях голодания субстратом для получения энергии могут стать кетоновые тела, свободные жирные кислоты.
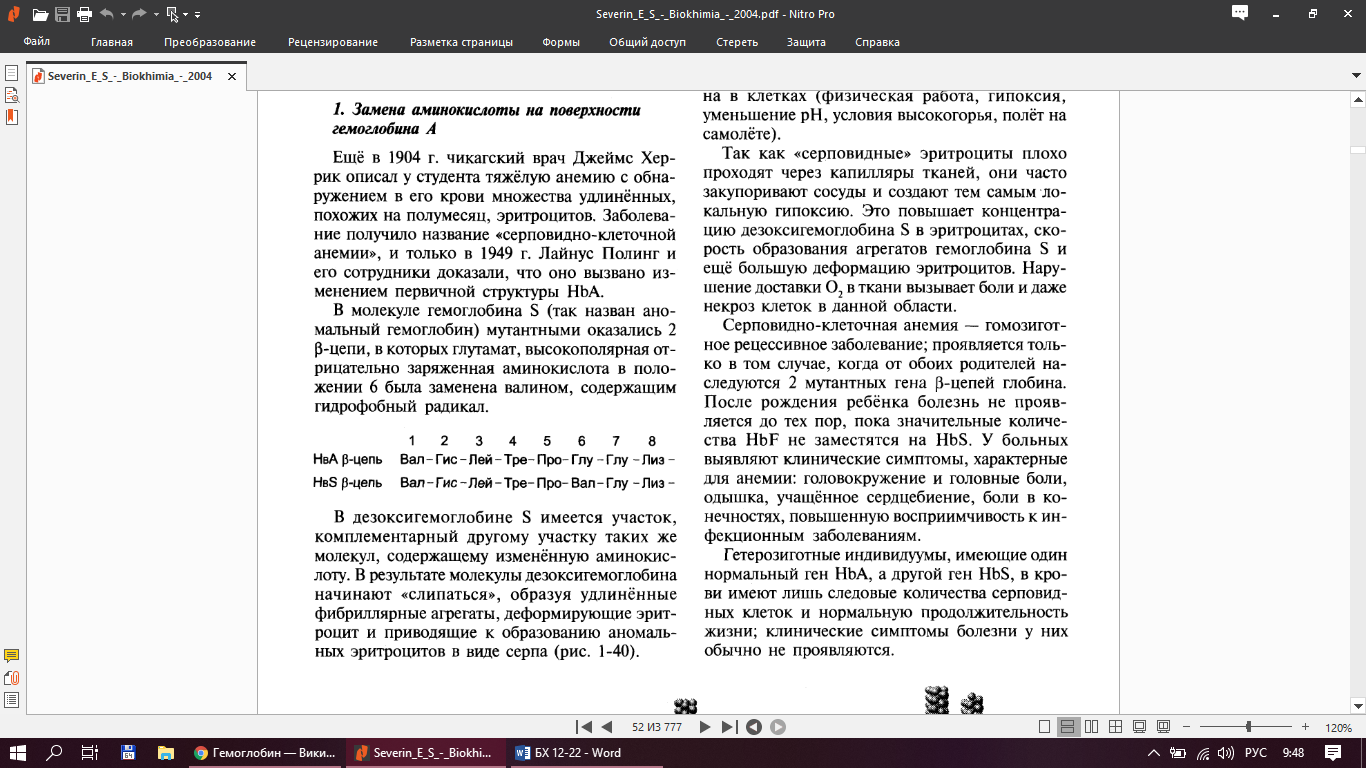
2.Гемоглобин Гемоглобин – белок, находящийся в эритроцитах человека.
Функции: 1. Перенос О2 из легких к периферическими тканям;
Участие в переносе СО2 и протонов из периферических тканей в легкие для последующего выведения из организма.
Структура:
Типы Hb:Hb А – основной гемоглобин взрослого организма, 98% от общего Hb, тетрамер, состоит из 2з полипептидных цепей α и 2 β (2α2β)
Hb А2– в организме содержится в меньшей концентрации 2% от общего Hb. 2α- и 2β-целей.
Hb АLe –гемоглобин А, модифицированный ковалентным присоединением к нему Глюкозы (так называемый гликозилированный гемоглобин)
Эмбриональный Hb–синтезируетсяв эмбрион-м желточном мешке через -2 нед после оплодотворения. Через 2 нед после формирования печени плода в ней начинает синтез-ся HbF, кот к 6 мес замещает эмбриональный.
HbF– фетальный гемоглобин, синтез-ся в печени и костном мозге плода до периода его рождения. После рождения замещается на HbA, кот начинает ситез-ся в клетках костного мозга уже на 8-м мес развития плода.
Кооперативное взаимодействие — взаимовлияние протомеров олигомерного белка друг на друга.
В легкихтакое взаимодействие субъединиц гемоглобина повышает его сродство к кислороду и ускоряет присоединение кислорода в 300 раз. В тканях идет обратный процесс, сродство снижается и ускорение отдачи кислорода также 300-кратное.
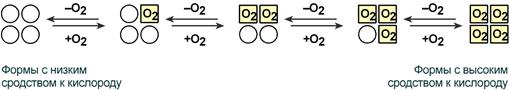
Схема кооперативного взаимодействия субъединиц гемоглобина
Объясняется такой феномен тем, что в легких при присоединении первой молекулы кислорода к железу (за счет 6-й координационной связи) атом железа втягивается в плоскость гема, кислород остается вне плоскости. Это вызывает перемещение участка белковой цепи и изменение конформации первого протомера. Такой измененный протомер влияет на другие субъединицы и облегчает связывание кислорода со второй субъединицей и тд.
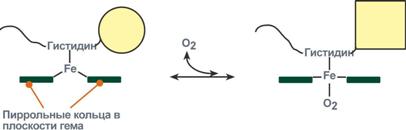
Влияние рН на сродство гемоглобина к кислороду носит название эффекта Бора.
При повышении концентрации протонов (закисление среды) в тканях возрастает освобождение O2 из оксигемоглобина. В легких после удаления угольной кислоты (в виде СО2) из крови и одновременном увеличении концентрации О2 высвобождаются ионы Н+ из Hb.
Изменение сродства гемоглобина к кислороду в тканях и в легких при изменении концентрации ионов H+ и О2 обусловлено конформационными перестройками глобиновой части молекулы. В тканях молекула О2 отрывается от железа и ионы водорода присоединяются к остаткам гистидина (глобиновой части), образуя восстановленный гемоглобин (H-Hb) с низким сродством к кислороду. В легких поступающий в больших количествах кислород "вытесняет" ион водорода из связи с остатком гистидина гемоглобиновой молекулы.
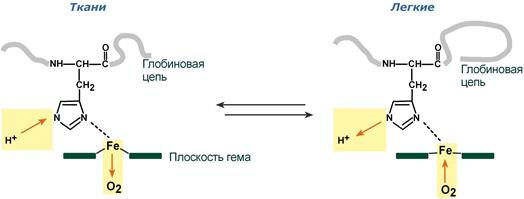
Механизм эффекта Бора:
2,3-Дифосфоглицерат образуется в эритроцитах из 1,3-дифосфоглицерата, промежуточного метаболита гликолиза, в реакциях, получивших название шунт Раппопорта
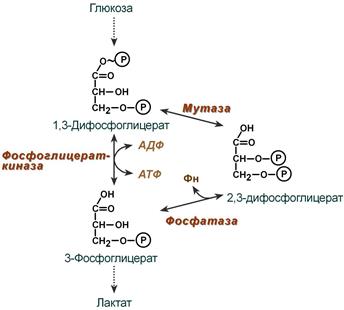
Реакции шунта Рапппорта:
2,3-Дифосфоглицерат располагается в центральной полости тетрамера дезоксигемоглобина и связывается с β-цепями, образуя поперечный солевой мостик между атомами кислорода 2,3-дифосфоглицерата и аминогруппами концевого валина обеих β-цепей, также аминогруппами радикалов лизина и гистидина.
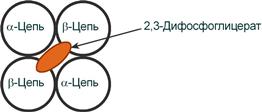
Расположение 2,3-дифосфоглицерата в гемоглобине :
Функция 2,3-дифосфоглицерата — в снижении сродства гемоглобина к кислороду, что имеет особенное значение при подъеме на высоту и при нехватке кислорода во вдыхаемом воздухе.
В этих условиях связывание кислорода с гемоглобином в легких не нарушается, так как концентрация его относительно высока. Однако в тканях за счет 2,3-дифосфоглицерата отдача кислорода возрастает в 2 раза.
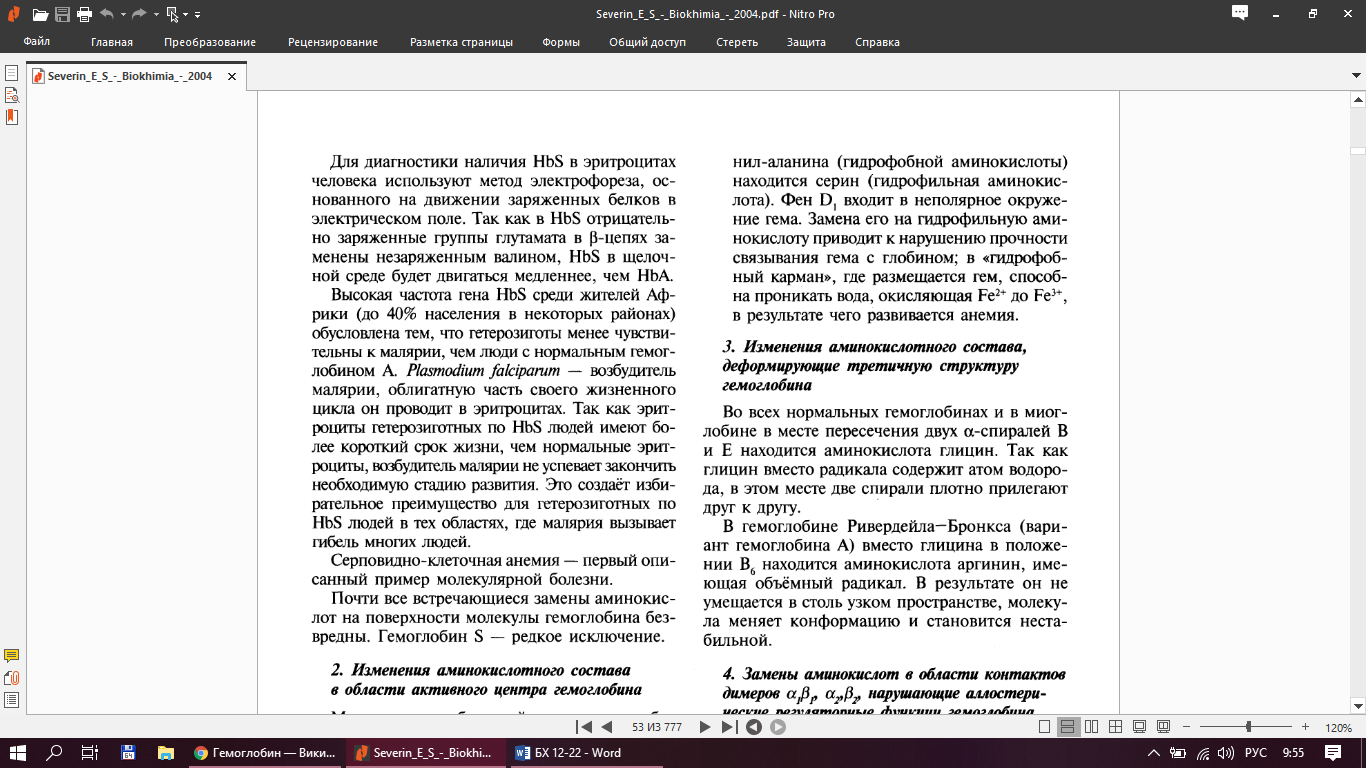
Гемоглобинопании - наследственные нарушенные первичной структуры и ф-ций гемоглобина А.
С
 ерповидно-клеточная
анемия (просто
прочитать)
ерповидно-клеточная
анемия (просто
прочитать)
Hb М – мутация в гене α- и β-цепи замена гистидина → тирозином. В результатеFe2+ окисляется в Fe3+ —называют метгемоглобином, отсюда и название HbM. У гетерозигот (цианоз, сязан с наруш. транспорта O2) У гомозигот – летальный исход.
Гемоглобин Хаммерсмита – в положении аланина (гидрофоб АК) находится серин (гидрофильн АК) →это приводит к наруш прочности связывания гема с глобином; в «гидрофобный карман», гле наход-ся гем проникает Н2О и окисляет Fe2+ до Fe3+→развивается анемия.
HbРивердейла–Бронкса – вместо глицина → аргинин (имеющий объемный радикал). В результате он не умещается в столь узком пространстве, молекула меняет конформацию и становится не стабильной.
Hb Кемпси – в β-цепи аспарагиновая к-та (участвующая в образ. Н-связей) → заменена на аспарагин. В результате Н-связи не образуются и наруш. стабильность конформации дезоксигемоглобина и сродство гемоглобина к О2 повышается → развивается анемия с выраженным цианозом.
Железодефицитнаяанемия—гематологическийсиндром, характеризующийсянарушениемсинтезагемоглобинавследствиедефицитажелезаипроявляющийся анемией.
Основные причины:
недостаток его в пище (несбалансированное вегетарианство),
заболевания ЖКТ со снижением всасывания (гипоацидные гастриты и энтериты),
потери железа с кровью при менструальных, кишечных или иных кровотечениях,
у новорожденных и грудных детей недостаток железа связан в первую очередь с недополучением его при внутриутробном развитии,
в связи с ускоренным ростом в первый год жизни (физиологическая анемия)
Недостаточный синтез цитохромов, железосодержащих белков и нарушение доставки кислорода к тканям (при снижении содержания гемоглобина) вызывает ряд специфических и неспецифических симптомов:
ухудшение внимания и памяти у детей и взрослых,
уплощение, волнистость и ломкость ногтей, появление исчерченности, белых пятен и полосок на ногтях,
выпадающий и секущийся волос,
поражение эпителия, проявляющееся в сухости и трещинах кожи рук и ног,
извращение обонятельных предпочтений – нравится запах краски, бензина, выхлопных газов, резины, мочи,
извращение вкусовых предпочтений – больные едят мел, штукатурку, уголь, песок, мясной фарш, лед.
3.Синдром дыхательных расстройств Задача про холестерин
Билет 2 1.Пути транспорта аммиака Для удаления аммиака из организма используется включение его в состав мочевины в печени и выведение ее с мочой, и удаление почками в виде аммонийных солей.
Однако, так как аммиак является чрезвычайно токсичным соединением, то предварительно в тканях (!) происходят реакции его обезвреживания (временного связывания) для переноса в печень и почки.
Пути:
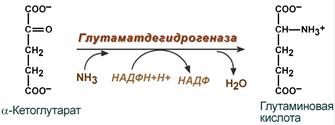
Синтез глутаминовой кислоты (восстановительное аминирование)– взаимодействие α-кетоглутарата с аммиаком. В качестве кофермента используется НАДФН. Происходит практически во всех тканях, кроме мышечной, но имеет небольшое значение, т.к. для глутаматдегидрогеназы предпочтительным субстратом является глутаминовая кислота и равновесие реакции сдвинуто в сторону α-кетоглутарата.
Синтез глутамина– взаимодействие глутамата с аммиаком. Является главным способом уборки аммиака, наиболее активно происходит в нервной и мышечной тканях, в почках, сетчатке глаза, печени. Реакция протекает в митохондриях.
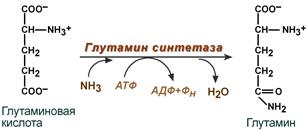
Так как глутамин проникает через клеточные мембраны путем облегченной диффузии, то он легко попадает не только в гепатоциты, но и в другие клетки, где есть потребность в аминогруппах. Азот, переносимый глутамином, используется клетками для синтеза пуринового кольца и гуанозинмонофосфата (ГМФ), глюкозамино-6-фосфата (предшественник всех остальных аминосахаров).
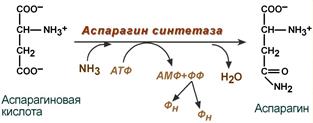
Синтез аспарагина – взаимодействие аспартата с аммиаком. Является второстепенным способом уборки аммиака, энергетически невыгоден, т.к. при этом тратятся 2 макроэргические связи.
Транспорт аммиака
Транспортными формами аммиака из тканей в печень являются глутамин и аланин, некоторое количество аммиака находится в крови в свободном виде.
Большая часть глутамина поступает от мышц и нервной ткани, аланин переносит аминный азот от мышц и стенки кишечника.
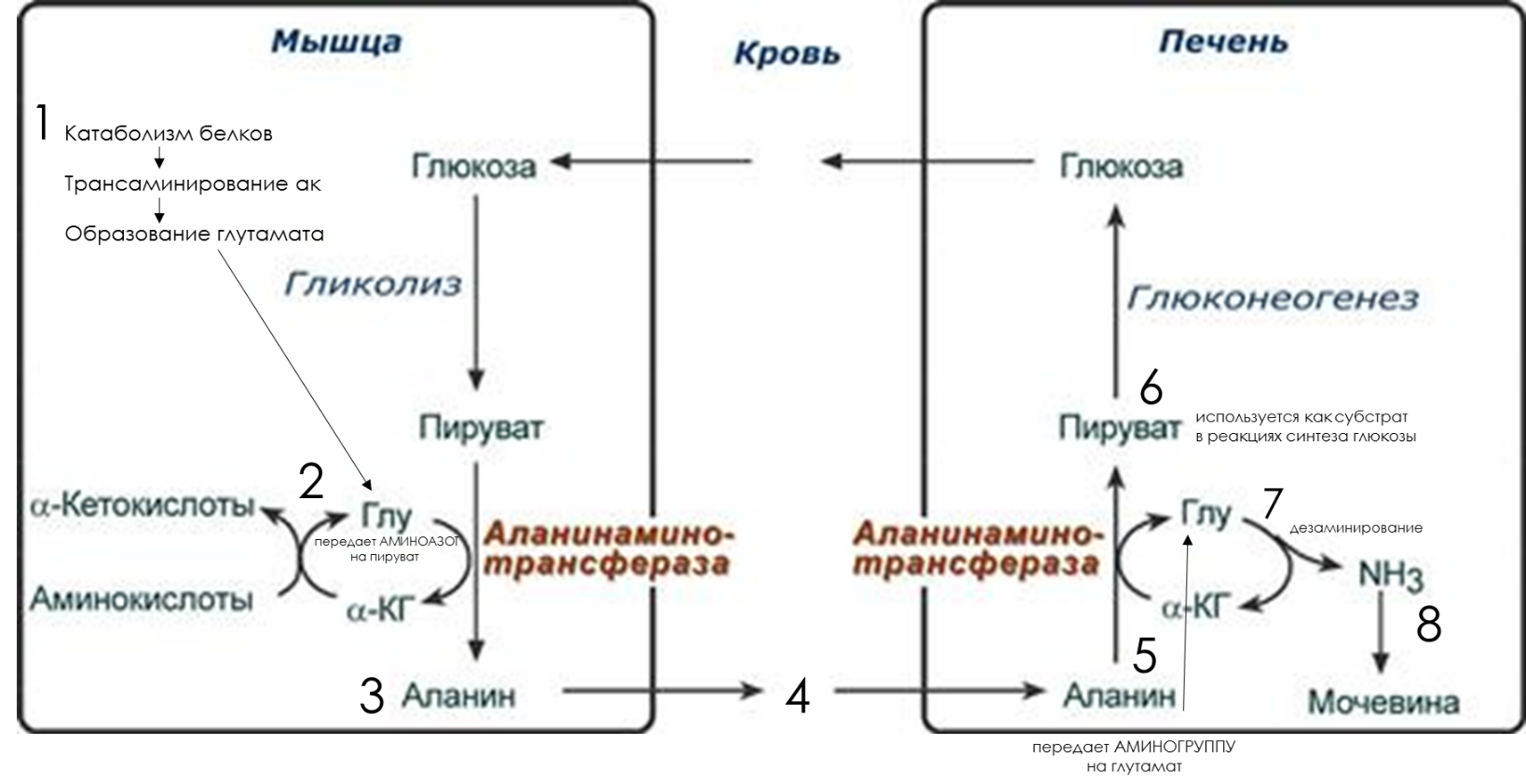
Глюкозо-аланиновый и глутаминовый цикл:
ЭТО РАСШИФРОВКА СХЕМЫ. ЭТО МОЖНО НЕ ПИСАТЬ, НО НУЖНО ЗАПОМНИТЬ.
При катаболизме белков в мышцах происходят реакции трансаминирования аминокислот, образуется глутамат (1), который далее передает аминоазот на пируват (2) и образуется аланин (3). Из мышц с кровью (4) аланин переносится в печень, где в обратной реакции передает свою аминогруппу на глутамат (5). Образующийся пируват используется как субстрат в реакциях синтеза глюкозы (глюконеогенез) (6), а глутаминовая кислота дезаминируется (7) и аммиак используется в синтезе мочевины (8).
Целевыми органами для транспорта аммиака являются печень, почки и кишечник.
В печени:
аспарагин и глутамин дезаминируются соответственно аспарагиназой и глутаминазой, образующийся аммиак используется для синтеза мочевины ,
аланин вступает в реакции трансаминирования с α-кетоглутаратом,
глутаминовая кислота подвергается окислительному дезаминированию.
В кишечнике часть глутамина дезаминируется глутаминазой. После этого образованный аммиак выделяется в просвет кишечника (не более 5%) или через кровь воротной вены уходит в печень, а глутамат вступает в трансаминирование с пируватом, в результате чего аминоазот переходит на аланин и с ним также поступает в печень;
В почках идет образование аммонийных солей с использованием глутамата, глутамина и аспарагина.
Белковый обмен
Больше половины синтезируемого за сутки в организме белка приходится на печень. Скорость обновления всех белков печени составляет 7 суток, тогда как в других органах эта величина соответствует 17 суткам и более.
К ним относятся не только белки собственно гепатоцитов, но и:
1) Идущие на "экспорт", составляющие понятие "белки крови"– альбумины, многие глобулины, ферменты крови (протромбин, церулоплазмин), а также фибриноген и факторы свертывания крови.
2) Аминокислоты подвергаются катаболическим реакциям с трансаминированием и дезаминированием, декарбоксилированию с образованием биогенных аминов (гистамин, гепарин).
3) Происходят реакции синтеза холина и креатина благодаря переносу метильной группы от аденозилметионина.
4) В печени идет утилизация избыточного азота и включение его в состав мочевины.
2.Факторы свертывания. Внешний и внутренний пути свёртывания Плазменные факторы свертывания(писать римскими цифрами)
1 фактор-фибриноген-гликопротеид, синтезируется в печени, нормальное содержание крови 2-4 г/л
2 фактор- протромбин (витамин К-зависимый и Са-зависимый) активируется Xа
3 фактор – тканевой тромбопластин – липопротеид, является фрагментом кл мембраны, поступающей в кровь при разрушении эндотелия клеток крови
4 фактор-ионы Са-необходимы для прикрепления 7а, 9а, 10а к фрагменту мембраны рузрушенного тромбоцита, также является активатором ферментативной активности 2а, 7а, 9а, 10а, без Са кровь не сворачивается! 5 фактор - проакцелерин. Активируется сначала тромбопластиногеном, а затем Са, является параферментоми при его отсутствии развивается парагемофилия 7 фактор –проконвертин(витамин К-зависимый и Са-зависимый)
8 фактор – состоит из 2 субъединиц
антигемофильный глобулин А(врожденный недостаток приводит к развитию гемофилии А)
фактор Виллибранда –за счет него происходит адгезия тромбоцита на субэндотелий
9 фактор – антигемофильный глобулин В или факток Кристмаса(витамин К-зависимый и Са-зависимый) Недостаток приводит к развитию гемофилии В
10 фактор – фактор Стюарта-Прауэра(витамин К-зависимый и Са-зависимый)
11 фактор – фактор Штейна (плазменный предшественник тромбопластина)Недостаток приводит к развитию гемофилии С
12 фактор – фактор Хагемана. Активируется при контакте с чужеродной поверхностью или измененным эндотелием, каллекерином в присутствии высокомолекулярного кининогена превращаясь в 12а. При недостатке - болезнь Хагемана
13 фактор – фибриностабилизующий фактор. Обеспечивает образование фибрин – полимера
14 фактор – прекалликреин или фактор Флетчера. Активируясь образует каллекреин
15 фактор – высокомолекулярный кининоген (фактор Фитцджеральда)
Роль Са и витамина К в свертывании Факторы свертывания крови (II, VII, IX, X) содержат в своем составе остатки γ-карбоксиглутаминовой кислоты, которые, посредством ионов Са++, обеспечивают связь этих ферментов с фосфолипидами клеточных мембран. В отсутствии ионов Са++ кровь не свертывается.
γ-карбоксилирование глутаминовой кислоты катализируется карбоксилазой, коферментом которой является витамин К. В связи с этим при недостатке витамина К нарушается γ-карбоксилирование выше названных факторов свертывания, что сопровождается кровоточивостью, подкожными и внутренними кровоизлияниями.
Внешний путь свертывания – нужны факторы, которые есть в тканях окружающих сосудов и попадают в крови только при повреждении тканей и сосудистой сетки

Внутренний путь свертывания – нужны факторы, которые есть в самой плазме крови и фосфолипидов тромбоцитов. Происходит активация 7 фактора в результате контакта с чужеродной поверхностью и калликреином в присутствии высокомолекулярного кининоген, которые адсорбируются на этой чужеродной поверхностью.