

5 Курс / Ревматология / рекомендации-Синдром-Альпорта
.pdfРазработчик:
Ассоциациянефрологов
МООТв« объединениерческоедетскихнефрологов» ОСП-Научно-исследовательскийклиническийинститутпедиа рии
ГБОУВПОРНИМУим...ПирогМинздраваРоссии
Клинические рекомендации «Д иагностика илечени е синдрома Альпорта удетей »
«Утверждено»
18декабря2014г.
2014г.Москва
Рабочаягруппа |
|
: |
|
|
ДлинВ.. |
– |
Заместительдиректорапонаучнработей |
ОСП-Научно- |
|
исследовательскийклиническийинститутпедиаГБОУВПОРНИМУрии |
|
|
||
им.Н.И.ПироговаМинздрав |
аРоссии ,доктормедицинскихнаук,профессор |
|
|
|
ИгнатоваМ.С. |
|
– почетныйпрезидентМООТв“ объединениерческое |
|
|
детскихнефрологов” |
, д.м.н.профессор, |
|
|
|
КоньковаН.Е. |
|
– ОСП-Научно-исследовательскийклиническийинститут |
|
|
педиатрГБОУВПОРНИМУи.м..иПирогова |
МинздраваРоссии |
, |
||
нефролог, |
к.м.н. |
|
|
|

Метоценкидикасилырекомендацийуровняихпредсказательности, использоваприсостд клининныхнаяерекомендацийческих
Посилерекомендацииподразделяютсянатрикатегориивубывающпор:уровдке1 мнь (экспертырекомендуют);уровень2эксп(предлагают);ртынедифференцированный« уровень» ( табл. 1 )Сила. предсказатрекомподренальнур4дацийзделенаовнясти
(табл. 2 ).
Таблица1Оцен. силырекомендаций
Оцрекомендацийнка
Уровень
Уровень1 «Эксперты
рекомендуют»
Уровень2 «Эксперты полагают»
«Нет градации»
(НГ)
Состороны пациентов
Подавляющее
большинство пациентов, оказавшихся подобнойситуации, предпочлибы следовать рекомендуемым путемилишь небольшаячастьиз нихотверглибыэтот путь
Большаячасть пациентов, оказавшихся подобнойситуации, выскбызтоа, зались чтобыследовать рекомендуемым путем,однако значительнаяасть отверглабыэтотпуть
Состоврачаоны
Подавляющему большсвоинству х пациентовврачбудет рекомслендовать именноэтимпутем
Дляразныхпациентов следуетподбирать различныеварианты рекомендаций, подходящиеименно. Каждомупациенту необходимапомощьв выбореипринятии решения,котороебудет соответствовать ценностями преданногопочтениям пациента
Данныйуровеньприменяетсятехслучаях,когдавоснову рекоменуклаздравыйсмыываетсяацииисследователя тогда,кобсуждаемаягдатеманедопусадепримененияквааетного системыдоказате,исповклиническойльзуемыхствпрактике.
*.
Дальнейшее
направление
использования
Рекомендацияможет бытьпринятав качествестандарта действия медицинского персоналав большинстве клиническихситуаций
Рекомендации, вер,потребуютятно обсуждения учавстиемех
заинтересованных сторондоприхнятия вкачестве клинического стандарта
-экспертаили
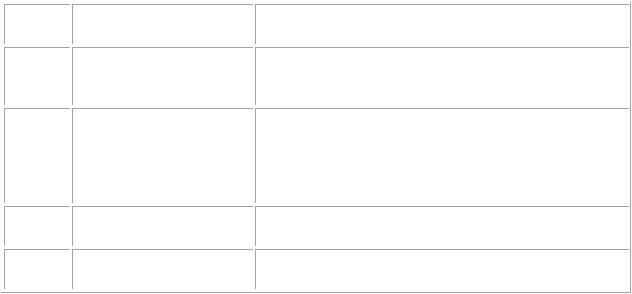
ТаблицаПредикт2 уровнирекомендацийрные
Уровень Характеристикауровня предсказательности
АВысокий
ВСредний
С |
Низкий |
D |
Оченьнизкий |
Значение/описание |
|
Экспертыабсолюуверетом,чтоприныо |
|
выполрекомендацииииданной, аблюдаемый |
|
эффектпочтиполностьюсовпадетожидаемым. |
|
Экспертыожидают,чтоприв полданненииой |
|
рекомендации,наблюдаемыйэффектскореевсего |
|
будетблизокожида,однаисемколючаетсяму |
|
возмтого,чтоонжносбудесущественноь |
|
отличатьсянего. |
|
Предсказываемыйэффектможетсущественн |
о |
отличреа. лтьсяного |
|
Предсказаэффектакрайнениночиед ньжно |
|
частобудетотличреа. лтьсяного |
|
РазделВведенгенетика1. , ,эп демиология,п тогенез |
|
|
|
|
|
|
|
||
Рекомендация1.1. |
СиндромАльпортаСА,(си:наследствоним |
|
|
енныйнефрит) |
– |
||||
неиммуннаягенетическидетерминир |
|
ованнаягломер,обусловленнаямутацлопатией |
|
|
|
|
|||
генов,кодирующихколлагентипабазальных4 мембран,проявляющаясягематурией/или |
|
|
|
|
|
|
|||
протеинурией,прогрессирующимсниженипочечныхфу, кцийсочредко |
|
|
|
|
|
етающаяся |
|||
патологиейслухазрения |
|
|
(НГ). |
|
|
|
|
|
|
ПриСАтакжередконаблюдается |
|
ейомиоматоз иизмененные тромбоциты. |
|
|
|||||
КоллагентипаIV |
|
состоитизгетер, оставленныхтримизкомбинаций6ров |
|
|
|
-тиальфа |
|||
цепей: |
|
|
|
|
|
|
|
|
|
o альфа3,ицеп54 |
и в гломерубазамембранахльныхярных( |
|
ГБМ); |
|
|
||||
o альфаи1цепив5 |
базальноймембране( |
БМ)капсБоиудистальнойлымена |
|
|
|
||||
тубулярнойБМ |
. |
|
|
|
|
|
|
||
Гены COL4A1и |
COL4A2 расположенына13х |
ромосоме, COL4A3и |
COL4A4 – на |
||||||
2 хромосоме, COL4A5и COL4A6 – на X хромосоме |
|
|
|
|
|||||
Рекомендация1.2. |
СиндромАльпорта |
– этиологетерогическинасленное |
|
дственное |
|||||
заболеваниемоногеннойприроды.Причиназаболеваниялежитмутацииодногоиз |
|
|
|
|
|
|
|||
генов: COL4A5,ПриклассическомCOL4A4,вариантеСАмутацияпроиCOL4A3. |
|
|
|
|
|
сходитв |
|||
генеCOL4A5,распонадложенномплечеХинном |
|
|
-хромосомыХ( |
q22.2),который |
|||||
кодируета5 |
-цепьколлагенатипаIV |
(НГ). |
Аутосомно-рецессивнаяформасиндрома |
|
|
||||
АльпортаобусловленамутациейгеновC0L4A3COL4A4,располнахроженныхмосоме |
|
|
|
|
|
|
|||
2икодирующихсоответственноа3 |
|
- иа4 -цепиколлагенаэтипаого. |
|
|
Аутосомно- |
||||
доминантнаяформаСА |
|
сцепгеннымленаокусомCOL4A3 |
|
-COL4A4. |
|
|
|
||
ЧастСАвпопуляциисостав1:5000Онпричинойяетужит.1%всехлучаев |
|
|
|
|
|
|
|||
хронипочнедостаточностиской( |
|
ХПН) вЕвропе. |
В 2,3%случапочевчн |
|
ая |
||||
трансплантация проводитсябольным |
СА. СА описанупредвсехраставителейнавсех |
|
|
|
|||||
континент.Встречаетсях |
|
СА очевидночаще,чемсообщм,всвязираетсязличной |
|
|
|
|
|||
пенетрантностьюиэкспрге,мутациянассивнкотегорогобуславливаетстью.Частота |
|
|
|
|
|
||||
различныхвариСАаутосомнонтов( |
|
|
-доминантного |
аутосомно-рецессивного) |
|||||
неодинаковара |
|
зличныхпопуляциях.Поэпидемиологическимданным |
|
|
вРоссии |
частота |
|||
СА средидетскпопуляциисоставлялай17:100000н |
|
|
аселен.Придлиятельном |
|
|||||
наблюдзачленамисемьинииСАбылоотмечено |
|
|
,чтоуженщинпожилоговозра |
|
|
ста, как |
|||
иупредставителеймужскогопола |
|
|
, отмечаетсяснижение функциипочек. |
|
|
|
|||
Рекомендация1.3. |
Генетическаядиагностикаважна,таккак |
|
отхарактераитипа |
|
|||||
мутацзавискл проявленияническиетзабол,скованиястьгрессирования |
|
|
|
|
|
|
|||
заболевивозранияст |
|
|
формитерхрониминальнойованияпочечнской |
|
|
|
|
||
недостаточностиХПН() |
(1А). |
|
|
|
|
|
|
||
КлассификацияСАвнастоящеевремяпроводитсяпотипунаследования:
•Х-сцеплендомиклассический( нантный);
•аутосомно-рецессивный;
•аутосомно-доминантный.
Около80%впопуляции |
состпациентывляютс |
Х-сцепленным доминантным |
|
вариантомСА,около15% |
– с аутосомно-рецессивнымСАиоколо5% |
– с аутосомно- |
|
доминантнымСА . |
COL4A5 выдеботяжелыеееяютварианты:делеции,нонсенсил |
|
|
Примутации |
|
|
|
сплайсингмутацииболеелегкийвариант |
– миссенсмутация. |
|
|
Улицмужскогопола |
Х-сцепленным доминантнымвариСАзаболеваниентом |
||||
имеетпрогрте:чтссирующеерминальнразвиваетсяниеХПН 50%возрастедо25 |
|
|
|
||
лет,у90% |
– ввозрастедо40летипочтиу100%ввозрастедо60лет.Приделеции |
|
или |
||
нонсенс-мутации COL4A5 рискразвитиятерминальнойХПНвозрастедолет30 |
|
|
|||
составляет90%,присплайсингмутации |
– 70%, априм мутацииссенс |
– 50%. Важность |
|||
определенобосновываетсятипамутацивыраженнойифенотип |
|
|
-генотипической |
||
корреляцией,чтоваж |
нодля прогнозирования сроковоказания |
помощизаместительная( |
|||
терапия)этимпациентам.ВбольшинствеслучсрокиаевзтерминальнойитияХПНу |
|
|
|
||
мальчиковс |
|
Х-сцепленным доминантнымвариантомСАсхожидругимичленамисемьи |
|
||
мужскпола,чтого |
|
позволяетпрог |
нозироватьвремянас ермиуплеХПНнияальной |
|
|
дажебезопределениягенотипа |
. Уженщинс |
Х-сцепленным доминантнымвариантомСА |
|||
такойвзаимосвязинаступлениясроковтермиХПНненаблюдаетсяальной,вероятно,из |
|
|
- |
||
заХ -инактив.Решениевопросаобактциивной |
|
ерапииуженщинс |
Х-сцепленным |
||
доминантнымвариСАзависитонфакторовомрискапрогрессирования:протеинурия, |
|
|
|
|||
макрогематуриянейросенстугоухость. рная |
|
|
|
|
|
|
Аутосомно-рецессивный |
САсвязанмутацобоихаллеляхей |
|
COL4A3или |
|||
COL4A4Генофенотипические. |
орреляциинетаквыражены,характерноразвитие |
|
|
|||
терминальнойХПНк30 |
-летнемувозрасту. |
|
|
|
||
Аутосомно-доминантСАсвязамутациейвный |
|
COL4A3илиCOL4A4 |
и |
|||
прогрессируетмедленномалопоказанийдляначалатерапиивдетскомвозрасте |
|
|
|
. |
||
Раздел2Клиническая. |
диагностика |
|
|
|
||
Рекомендация2.1. |
ДлядиагСАнеобходостпритрехсутствиекиизпятимо |
|
|
|
||
признаков:гематуриялетальныйисходотХПНвсемье;гематурия/или |
|
|
|
|
||
протеинуриявсемье;специфичизменБМклуеубонияскиеприльногочков |
|
|
электронной |
|||
микроскопии( |
ЭМ) |
биоптата;снижениеслухподаннымудиометрического |
|
|
||
исследования;врожденпатологиязре ая |
|
|
.Этикробосновываюттерии |
|
||
необходимтщательногосборада стьаличииныходнклиническойтипнойкартины |
|
|
|
|
||
илетальныхисходовотХП |
|
|
Нукровныхродственников,провэлектроннодение |
|
- |
|
микросслкопическн дованфробиоптатбследование,тщательноеяго |
|
|
пробандаи |
|||
родственников дляисключениянейросенсорнойтугоухости |
ипатологиизрения |
(наиболее |
||||
характерноналентиконусаичие) |
|
(НГ) . |
|
|
|
|
Ув сехбольныхимеетсябессимптомаленькихмикрогематурия;девочек |
|
|
|
|||
мальчиковонаможетвозникатьпериодич.Примерно50%случачерски1езв |
|
|
|
-2дня |
||
после респираторной инфекциипоявляетсямакрогема.Умальчиковчастурия |
|
|
|
|||
наблюдаетсяпротеинурия,но |
|
девочеконаможетбытьслабой,преходящейиливообще |
|
|
||
отсутств.Ковторомудесятилетиюватьжизнипротеинуобычнопрогрессирует, ия |
|
|
|
|
||
преимущелицмужпола,скоготвенно |
|
нередкопревышаетг/ 1 |
24ч |
иможет |
||
сопровождатьсянефротическимсиндромом. |
|
|
|
|
|
|
Квнепочечным |
проявлениям СА относятсятугоухостьинарушениязрения. |
|
|
|||
Двустонейроглухота,сенняякотораясорнаяикогданепроявляетсяранниесроки |
|
|
|
|
||
послерождения,отмечаетсяу90%гемизиготныхмальчиковсХ |
|
|
|
-сцепленнымсиндромом |
|
|
Альпорта,у 10%гетерозиготныхдево |
|
чексХ |
-сцепленным |
СА иу 67%больныхс |
|
|
аутосомно-рецессивнымсиндромом.Вначаленарушаетсявосприятиезвуковвысокой |
|
|
|
|||
част,нопостепетыбольныеперестаютслышанообычнуюзговорнуюречь,иим |
|
|
|
|
||
приходитсяпользоватьсяслуховымаппаратом.Глазныенаруше |
|
|
|
ния выявляются у30 -40% |
||
больныхсХ |
-сцепленным СА и включаютпередннтсмещение( цекой тральнойус |
|
|
|||
частихрусталивпереднююкамеглаза), раунажелтомпинкипятнеэрозии |
|
|
|
|
||
роговицы.Вредкихслучаяхнаблюдается |
|
лейомиоматоз пищевода,трахеи,бронхо |
ви |
|||
женскихнаружныхполовыхорг,такжеизменениеновтромбоцитов. |
|
|
Лейомиоматозом |
|||
называетсяпатологическийпроцесс,хара теристикамиоторогоявляютсяпролиферация гладкихволоконмышц,тканяхиорганах,котс держатврыенормегладкомышечную ткань.
Рекомендация2.2. |
Приаутосомно |
-рецессивныхформСА,атакжечасти |
|
|
||||
случаеваутосомно |
-доминантных,втомч прислеХ |
|
|
-сцепленныхформахзаболевания, |
|
|||
этихпризнаковнедостаточнонеобхпр молведдимокулярноние |
|
|
|
-генетического |
||||
исследования (НГ). |
|
|
|
|
|
|
||
Рекомендация2.3. |
Электронно-микроскопическими |
критериямидиагностики |
|
|||||
являются: |
истБМ,ончсобенносреднейниепластинкиlaminaодновременноdensa, |
|
|
|
|
|||
наблюдарасщБМипоявеплтеселяение |
|
|
|
оистости. |
ПриЭМбиоптатапочки |
|
||
одновременностонкимиГБМвыявл |
|
|
яюутсяолще |
нныеГБМсучасткамипросветления, |
|
|||
напоминающиепчелинсо.БМтыеряструксво ,внунихпоявляютсяурури |
|
|
|
|
|
|||
скоплениятонкогранувещества.Помередалярного |
|
|
|
ьнейшегопрогрессированияболезни |
|
|||
происходиттяжелаядеструкцияГБМеедальне |
|
|
йшим утолщениемдистрофией |
(НГ). |
||||
Внормемоноклонаантитеальфа3 ьные |
|
|
-цепиколлагенатипаIV(MAB3) |
|
||||
моноклонаантитеальфа5 ьные |
|
-цепиколлагенатипаIV(MAB5)окрашиваютвсю |
|
|
||||
толщинуклубазальнойочковоймембраны,такжебазальнуюмембранудистальн |
|
|
|
|
ых |
|||
канальцев.ВсемьяхX |
|
|
-сцепленным СА окрашиваниенаMAB3MAB5отсутствует |
|
|
|||
мужчинноситпрерывистыйхарактеруженщин.Такуюкартинуможнонаблюдать60 |
|
|
|
|
- |
|||
70%семей.Убольныхприаутосомно |
|
-рецессивномтипе |
СА окрашиваниенаMAB5видно |
|
||||
набазальной |
|
мембрБоуменовойкапсулынеисобира |
|
|
тельных прото,тогдакаков |
|
||
окрашиваниенаMAB3отрицательное. |
|
|
|
|
|
|
||
ОкрашиваниеMAB3нормекоженевыявляется,тогда,какMAB5окрашивают |
|
|
|
|
|
|||
эпидербазальнуюмембрану.ЭтоокрашиванотсутствуетобычнопрXи |
|
|
|
|
- |
|||
сцепленном СА умужчин,тогдакакженщинвыявлясегментарнлокализтсяация |
|
|
|
|||||
антигена.Убольныхприаутосомно |
|
-рецессивномтипенаследованияокрашиваниеMAB5 |
|
|
||||
эпидермальнойбазальноймембраныявляетсянормальным. |
|
|
|
|
|
|
||
Рекомендация2.4. |
Гематуявляетсянаиболеерия |
|
аннимпризнакомСА. |
|
||||
Микроальбуминурия,впоследующемпротеинурия,выявля |
|
|
|
етсявболеепозднириодй |
|
|||
еевыражопрскоенностьделяетпрогрессированиязаболеванияь |
|
|
|
(НГ). |
|
|||
Рекомендация2.5. |
Интерстициальныйфиброз,какпридругихгломерулопатиях, |
|
|
|||||
играетважнуюрольпрогрессирСА.Поэтмерынапромуваниивленные |
|
|
|
|
|
|||
предупфореждениемированияпрогрессированинтерстицфиброзприСА ая льного |
|
|
|
|
|
|||
необходимоначинатьвдетскомвозрасте |
|
|
(НГ). |
|
|
|
||
РазделЛечение3. |
|
|
|
|
|
|
|
|
Рекомендация3.1. |
МальчикамсСА,связанным |
|
мутациейCOL4A5,которые |
|
||||
имделециюют,нонсенсилисплайсингмутациюилиимеютвсемьеслучаиразвития |
|
|
|
|
|
|||
терминальнХПНдолетнего30возрастад провйлжнаборанняялеедиться |
|
|
|
|
|
|||
активнаяераснижающаяпияротеинурию |
|
|
|
,чтопозволяетпредо вратить |
|
|||
повреждение эпителиальклетокпочечканихныальцевтрофиюхпредупреждает |
|
|
|
|||||
развитиеинтерстицифиброзального |
|
(НГ). |
|
|
|
|
||
Рекомендация3.2. |
Цельантипротеинуртерапротеинуриисниженической |
|
|
|||||
менее0,5мг/ креатиилина50%,еслиначиназначениельноепротеинур |
|
|
|
иименее1,0 |
||||
мг/ креатинина |
(НГ). |
|
|
|
|
|
|
|
Рекомендация3.3. ПервойлиниейантипротеинурическойтерапииСАявляются ингибиторыангиотензин -превращающего ферментаиАПФ()Используют. миприлв
дозе1 -2мг/ |
2/24ч,эналаприлвдозирвдввышеовке(2 |
|
|
|
|
-4мг/ |
2/24ч ), |
лизиноприл, |
|||
фозиноприл, |
квинаприл вдозировкевчетверовыше(4 |
|
|
-8мг/ |
2/24ч) (НГ). |
|
|
||||
Рекомендация3.4. |
ВторойлиниейантипротеинурическойтерапииСАявляются |
|
|
|
2/24ч, |
||||||
блокаторырецептангиБРА()Испров.тензиналользуютзартанвдозе12,5мг/ |
|
|
|
|
|
|
|||||
удваиваядозукаж |
|
|
дыемесяцадостижен3 максимальнойдозыпр(отсутствиия |
|
|
|
|
||||
побочныхэффектов) мг/50 |
|
2/24ч., |
ирбисартан – |
|
тройндозалозаяртана(37,5 |
|
|
||||
мг/ 2/24ч),вальсартан |
|
– 1,5дозалозартана |
|
(18,75 мг/ |
2/24ч)НГ)(. |
|
|
||||
Комментарии |
|
|
|
|
|
|
|
|
|
||
Осложнентерапи. ия |
|
Лечениепроводится |
|
подрегуляконтролемуровняным |
|
калия. |
|||||
Повышеегоуровможетбытьсвязаноиеядействием |
|
|
|
|
|
иАПФилиБРАтребует |
|
||||
снижедозыпрепниеа50%,априратаперсгиперкалстенцииемии |
|
|
|
|
|
– полнаяотмена |
|||||
терапии. |
|
|
|
|
|
|
|
|
|
|
|
ВтерминальнойстадииХПНвыполняютгемодиализили |
|
|
|
|
|
рансплантациюпочки. |
|||||
Примерноу больных5%,перенесшихтрансплапочки,развиваетсянефртац, ю |
|
|
|
|
|
|
|
||||
обусловлантителакбазальноймембраненныйклуби,главнымобразомчкову |
|
|
|
|
|
|
лиц |
||||
мужскогопола |
сХ |
-сцепленнымсиндромом,укоторыхХПНдостигаеттерминальной |
|
|
|
|
|||||
стадиидо30 |
|
-летнеговозраста. |
|
|
|
|
|
|
|
||
Раздел4. |
Втопрофилактикаичная |
. |
|
|
|
|
|
|
|||
ВсемьяхХ |
|
-сцепленнойформой |
|
|
СА приизвесмувозможнатацияхных |
|
|
||||
пренатальнаядиагностика |
|
,чтокрайневажно,есплодмужскогоипола |
|
|
|
. |
|
||||
Раздел 5.Перспектвтерапиивы |
СА. |
|
|
|
|
|
|
||||
Реальныхперспектив |
|
азработкиэтиотте апииной |
|
|
СА внастоящеевремянет. |
|
|
||||
Библиография |
|
|
|
1. ФокееваВ..Наследственныйнефрит. |
Вкн.: |
Детскаянефрология |
(руководстводля |
врачей)Под/ред.М.С.Игнатовой,Ю.Е.ВельтищеваЛ.Медицина: , 1989. |
|
|
-с.244 -256. |
2.Atkin C.L., Gregory V.S., Border W.A. Alport syndrome. In.: Diseases of the kidney /Ed. R.W. Schrier, C.M. Cottschalk.- 4th ed.-Boston:Little,1989.
3.Bekheimia M.R., Reed B., Gregory M.C. et al. Genotype-phenotype correlation in X-linked Alport syndrome. J.Am.Soc.Nephrol. 2010; 21: 876-883.
4.Gross O., Netzer K.O., Lamberty R. et al. Meta-analysis of genotype-phenotype correlation in X-linked Alport syndrome. Nephrol.Dial. Transp. 2002;17:1218-1227.
5.Grunfield J-P., Noel L.H., Hafez S, Droz D. Renal prognosis in women with hereditary nephritis. Clin Nephrol. 1985;23:267-271.
6.Jais J.P., Knebelmann B., Giatras I. et al. X-linked Alport syndrome: natural history in 195 families and genotype-phenotype correlations in males. J.Am.Soc.Nephrol. 2000; 11: 649657.
7.Jais J.P., Knebelmann B., Giatras I. et al. X-linked Alport syndrome: natural history and genotype-phenotype correlations in girls and womans belonging to 195 families: a “European Community Alport Syndrome Concerted Action” study. J.Am.Soc.Nephrol. 2003; 14: 2603-2610.
8.Pochet J.M., Bobrie G., Landais P. Et al. Renal prognosis in Alport’s and related syndromes: influence of the mode of inheritance. Nephrol.Dial. Transp. 1989;4:1016-1021.
9.Rheault M.N., Kren S.M., Hartich L.A. X-inactivation modifiesdisease severity in female carriers of murine X-linked Alport syndrome. Nephrol.Dial. Transp. 2010;25:764-769.
10.Rheault M.N. Women and Alport syndrome. Pediatr Nephrol. 2012; 27(1): 41-46.