

МИНИСТЕРСТВО НАУКИ И ВЫСШЕГО ОБРАЗОВАНИЯ РФ
Федеральное государственное автономное образовательное учреждение высшего образования
«ЮЖНЫЙ ФЕДЕРАЛЬНЫЙ УНИВЕРСИТЕТ»
Академия психологии и педагогики психологии
Кафедра психофизиологии и клинической психологии
КУРСОВАЯ РАБОТА
на тему:
Нейродегенеративные заболевания
Студентки 2 курса с-о-1 группы
очного отделения
Мурадалиевой Карины Салаудиновны
Проверила:
к.психол.н., доцент
Ковш Екатерина Михайловна
Ростов-на-Дону,
2021 г.
ОГЛАВЛЕНИЕ
|
стр. |
|||
Введение…………………………...………………………………………..... |
3 |
|||
Глава I. |
МЕДИЦИНСКИЕ АСПЕКТЫ НДЗ ……………………...... |
4 |
||
|
|
|
4 |
|
|
1.2. |
Болезнь Альцгеймера (этиология, патогенез, клиническая картина, лечение) ……………………… |
8 |
|
|
1.3.
1.4. |
Болезнь Паркинсона (этиология, патогенез, клиническая картина, лечение) ……………………… Диагностика и профилактика НДЗ…………………... |
16 23 |
|
Глава II. |
ПСИХОЛОГИЧЕСКОЕ СОПРОВОЖДЕНИЕ НДЗ |
29 |
||
Заключение |
……...………………………………………………………… |
32 |
||
Литература |
……...………………………………………………………… |
33 |
||
|
|
|
||
Введение
Нейродегенеративные заболевания (НДЗ)- это гетерогенная группа относительно медленно прогрессирующих наследственных или приобретенных фатальных патологий нервной системы. Общими признаками этих болезней являются нарушение моторики, памяти и формирование белковых агрегатов [Е.А. Никитина, 2015].
На данный момент от различных НДЗ страдают более 30 млн человек. Согласно прогнозам, количество заболевших будет только расти [Р.В. Магжанов, К.З. Бахтиярова, Е.В. Первушина, 2018]. Такое широкое распространение НДЗ связано, в первую очередь, с увеличением продолжительности жизни в развитых странах, так как процентная вероятность манифестации болезни увеличивается после 60-65 лет. Данная группа заболеваний влияет на все сферы деятельности человека. Вышеперечисленное делает НДЗ одним из основных приоритетов для исследования врачей, ученых-химиков, биологов и психологов.
Цель данной работы заключается в изучении и обобщении ряда вопросов, касающихся актуальной информации об НДЗ.
Задачи:
Описать этиологию, патогенез и клиническую картину наиболее часто встречающихся НДЗ;
Рассмотреть современные представления о диагностике, лечении и профилактике НДЗ;
Сформулировать на основе имеющихся данных возможности дальнейшего исследования.
Глава 1 медицинские аспекты ндз
Классификации НДЗ
Проблемы систематизации НДЗ возникают в связи с неясностью их этиологии и отдельных аспектов патогенеза. Основные виды классификации:
Клиническая (выделяется на основе патологической анатомии и клинических признаках) [Б. Браунвальд и др., 1997].
I. Расстройства, характеризующиеся прогрессирующей деменцией, при отсутствии других выраженных неврологических симптомов.
А. Болезнь Альцгеймера.
Б. Сенильная деменция альцгеймеровского типа.
В. Болезнь Пика (лобарная атрофия).
II. Синдромы прогрессирующей деменции, сочетающейся с другими выраженными неврологическими нарушениями.
А. Преимущественно взрослого возраста:
1. Болезнь Гентингтона
2. Множественная системная атрофия, сочетание деменции с атаксией и/или проявлениями болезни Паркинсона
3. Прогрессирующий супрануклеарный паралич (синдром Стила - Ричардсона - Ольшевского)
Б. Преимущественно детского и молодого взрослого возраста
1. Болезнь Галлервордена - Шпатца
2. Прогрессирующая семейная миоклонус-эпилепсия
III. Синдромы, сопровождающиеся постепенным развитием нарушений позы и движений
А. Дрожательный паралич (болезнь Паркинсона)
Б. Стрионигральная дегенерация
В. Прогрессирующий супрануклеарный паралич
Г. Торсионная дистония (торсионный спазм, деформирующая мышечная дистония)
Д. Спастическая кривошея и другие органические дискинезии
Е. Семейный тремор
Ж. Синдром Жилль де ла Туретта
IV. Синдромы, сопровождающиеся прогрессирующей атаксией
А. Мозжечковые дегенерации
1. Мозжечковая кортикальная дегенерация
2. Оливопонтоцеребеллярная атрофия (ОПЦА)
Б. Спиноцеребеллярные дегенерации (атаксия Фридрейха и сходные расстройства)
V. Синдром центральной недостаточности вегетативной нервной системы (синдром Шая - Дрейджера)
VI. Синдромы мышечной слабости и атрофий без нарушений чувствительности (болезни двигательного нейрона)
А. Боковой амиотрофический склероз
Б. Спинальные амиотрофии
1. Семейная спинальная амиотрофия детского возраста (болезнь Верднига - Гоффманна)
2. Юношеская спинальная амиотрофия (болезнь Вольфарта - Кугельберга - Веландер)
3. Другие формы семейных спинальных амиотрофий
В. Первичный боковой склероз
Г. Наследственная спастическая параплегия
VII. Синдромы сочетания мышечной слабости и атрофий с расстройствами чувствительности (прогрессирующие невральные амиотрофии, хронические семейные полиневропатии)
А. Перонеальная амиотрофия (Шарко - Мари - Тута)
Б. Гипертрофическая интерстициальная полиневропатия (Дежерина - Сотта гипертрофический неврит)
В. Различные формы хронической прогрессирующей невропатии
VIII. Синдромы прогрессирующей потери зрения
А. Пигментная дегенерация сетчатки (пигментный ретинит)
Б. Наследственная атрофия зрительных нервов (болезнь Лебера)
Молекулярная (сформирована на основе особенностей нарушений конформации белка) (рис 1.) [С.К. Евтушенко, 2015].
Таупатии:
Болезнь Альцгеймера
Прогрессирующий супрануклеарный парез взора
Кортикобазальная дегенерация
Болезнь серебряного зерна
Фронтотемпоральная деменция и паркинсонизм 17 хромосомы
Болезнь Пика
Синуклеопатии:
Болезнь Паркинсона
Деменция с тельцами Леви
Мультисистемная атрофия
Тринуклеотидные заболевания:
Хорея Геттингтона
Спинобульбарная мышечная атрофия, тип Кеннеди
Атаксия Фридрейха
Спиноцеребеллярная атаксия
Прионные заболевания:
Болезнь Крейтцфельдта — Якоба
Синдром Герстмана — Штраусслера — Шейнкера
Фатальная семейная бессонница
Куру
Заболевания мотонейрона:
Боковой амиотрофический склероз
Первичный боковой склероз
Спинальная мышечная атрофия
Нейроаксональные дистрофии:
Инфантильная нейроаксональная дистрофия
Нейродегенерация с отложением железа в мозге
Фузопатии:
Дегенерация фронтотемпоральных долей с FUS (FTLD-FUS)
Neuronal intermediate filament inclusion disease (NIFID)
Basophilic inclusion body disease (BIBD)
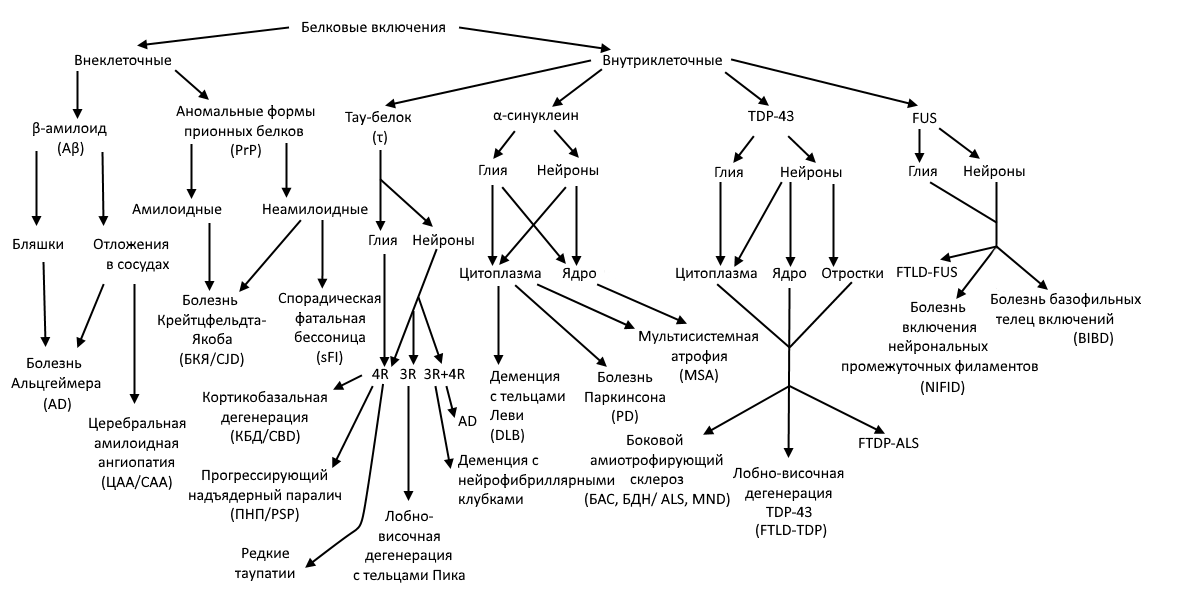
Рисунок 1, Классификация НДЗ, ассоциированных с протеинопатиями
Болезнь Альцгеймера
БОЛЕЗНЬ АЛЬЦГЕЙМЕРА (БА)- это нейродегенеративное заболевание, характеризующееся необратимой гибелью нейронов (преимущественно в областях гиппокампа, теменной и височной коры головного мозга), приводящей к прогрессирующим когнитивным нарушениям –деменции.
БА считается одним из наиболее часто встречающихся НДЗ и самой распространенной причиной деменции в развитых странах (от 50 до 70 процентов случаев) [М.В. Нестерова, 2018]. На 2010 г. в мире проживало около 33 млн больных, согласно прогнозам, их количество увеличится: к 2030 г. будет равно 66 млн, а к 2050 г. – 115 млн [В.А. Парфенов, 2015]. В связи с ростом заболеваемости, XXI в. считают веком эпидемии БА, [В.В. Захаров и др., 2015], [Е.Е. Васенина и др, 2013].
Согласно МКБ-10, выделяются следующие виды БА:
Болезнь Альцгеймера с ранним (до 65 лет) началом (тип 2 болезни Альцгеймера, пресенильная деменция альцгеймеровского типа). Это классическая («чистая») БА
Болезнь Альцгеймера с поздним (после 65 лет) началом (тип 1 болезни Альцгеймера, сенильная деменция альцгеймеровского типа).
Атипичная болезнь Альцгеймера (деменция смешанного типа)- сочетание признаков деменции при БА и деменции иной причины.
Существуют несколько клинических типов проявления БА: амнестический, афазический, апраксический, с визуально-пространственными нарушениями и поведенческий [Н.Н. Коберская, 2017].
Этиология.
БА- заболевание с многофакторной этиологией. Помимо влияния генетических мутаций, для возникновения патологического процесса необходимо воздействие определенных внешних и внутренних факторов. Семейные формы заболевания (генетически обусловленные) составляют 10% [М.В. Угрюмов, 2014]. Согласно современным исследованиям выделяют 4 гена, носительство которых вызывает высокую вероятность развития БА:
На 21 хромосоме ген, кодирующий предшественник амилоидного белка (β-АРР). Мутация в данном гене встречается у 3-5% всех пациентов с семейной формой пресенильного типа болезни. С β-АРР также связывают развитие деменции при синдроме Дауна [Н.Н. Яхно и др., 2011].
На 14 хромосоме пресенилин-1 (PSN-1). Наиболее частая мутация происходит в PSN-1. Носительство мутации в данном гене имеет полностью пенетрантный характер- вызывает развитие пресенильной формы БА почти в 100% случаев [Е.И. Гусев, 2018], [Н.Н. Яхно и др., 2011];
На 1 хромосоме пресенилин-2 (PSN-2). Мутации в данном гене редки, носят не полностью пенетрантный характер, влияют на развитие как ранних, так и поздних семейных форм заболевания [М.В. Угрюмов, 2014];
На 19 хромосоме- изоморфный вариант гена аполипопротеина Е (АроЕ4). Обуславливает возникновение поздней семейной и спорадической формы БА, увеличивает риск развития БА в 2 раза по сравнению со среднестатистическим [Н.Н. Яхно и др., 2011]. Считается, что изоформа ApoE способна влиять не только на агрегацию и клиренс Аβ в головном мозге, но и на синаптическую пластичность, клеточную сигнализацию, транспорт липидов и метаболизм, на нейровоспаление [D. M. Holtzman et. al., 2012].
Генетические факторы БА продолжают изучаться, так как заболевание возникает и тогда, когда перечисленные гены у человека не наблюдаются. Повлиять могут также экзогенные и эндогенные воздействия:
недостаток витамина D (ингибирует гамма-секретазу- фермент, участвующий в синтезе β-амилоидного пептида (βАП));
нарушения сна (в частности, влияет уменьшение длительности фазы медленного сна, так как именно во время нее происходит утилизация предшественников альцгеймеровского амилоидного белка- аминокислотных цепей абета-42);
нарушения обоняния (согласно исследованиям, депривация обонятельных стимулов ускоряла процесс амилоидогенеза) [В.В. Захаров и др., 2015].
Артериальная гипертензия, инсулинорезистентность, возрастной дефицит половых гормонов, гипотиреоз, печеночная и почечная недостаточность, гипоксия, церебральная ишемия, и нехватка витаминов группы В- все эти факторы также влияют на реализацию генетической программы [В.В. Захаров и др., 2015], [Н.Н. Яхно и др., 2011].
Патогенез.
Точные механизмы патогенеза БА еще не выяснены учеными, однако существует несколько гипотез, объясняющих течение заболевания.
Холинергическая гипотеза является наиболее ранней идеей. Согласно ей, основной механизм возникновения БА- недостаток ацетилхолина. В 1982 году было выяснено, что при НДЗ, ассоциированных с деменцией, возникает дефицит ацетилхолина. Это нейромедиатор, основной функцией которого является перевод информации из кратковременной в долговременную память. Позже установили, что патологический процесс при БА тесно связан с нарушениями в холинергической системе- самые ранние и грубые изменения при этом заболевании возникают в структурах гиппокампова круга и медиобазальной лобной коре. На основе этих тезисов была выстроена стратегия лечения ингибиторами холинестеразы, однако подобная практика не возымела успеха [Б. Браунвальд и др., 1997], [Л.А. Дзяк, Е.С. Цуркаленко, 2019]
Амилоидная гипотеза. Изменения мозга при БА на молекулярном уровне заключаются в нарушении процесса образования βАП из его предшественника. Предполагается, что в норме βАП является компонентом врожденного иммунитета [Stephanie J. Soscia, et. al., 2010]. Согласно амилоидной гипотезе, нарушение баланса между расщеплением и синтезом βАП, вызывает процесс, называемый конформационной перестройкой. В структуре пептида начинают преобладать β-тяжи. В результате перестройки βАП образует нитевидные амилоидные агрегаты- нерастворимые жесткие фибриллы больших размеров. Они токсичны и провоцируют разрушение окружающих клеток. Помимо этого, βАП переводит нетоксичный белок в патологическую конформацию. Этот процесс влечет за собой изменения на клеточном уровне- начинают образовываться амилоидные бляшки (локальные внеклеточные скопления βАП, окруженные микроглией, аксонами, дендритами в стадии дегенерации) [М.В. Угрюмов, 2014], [И.В Литвиненко и др., 2019].
Тау-гипотеза. Патологический амилоидоз тесно связан с процессом образования нейрофибриллярных клубков (НФК). Это внутриклеточные включения, состоящие из гиперфосфорилированного тау-белка. Функция этого протеина в норме- стабилизация микротрубочек. Гиперфосфорилирование приводит к нарушению его связей с микротрубочками, что влечет за собой гибель нейрона. Количество НФК коррелирует с тяжестью протекания БА, разрушением мозговых клеток, являясь маркером патологии [И.В Литвиненко и др., 2019].
В связи с вышеперечисленными изменениями, происходит постепенная атрофия мозговой ткани- погибает большое количество нейронов, разрушаются синапсы. Особенно заметны нарушения в гиппокампе, височной коре, базальных ядрах, поясной извилине, в медиобазальных областях коры больших полушарий. Масса и объем мозга больного заметно уменьшается - борозды углубляются, извилины сужаются, желудочки расширяются, толщина коры некоторых полей сужается. Развивается компенсаторная гидроцефалия- ликвор постепенно заполняет «пустоты» мозговых структур. [И.Н. Боголепова и др., 2019]
Клиническая картина.
Чаще всего БА манифестирует у людей 60-65 лет. Деменция альцгеймеровского типа характеризуется медленным малозаметным началом и необратимой прогрессией. Болезнь подразделяется на несколько стадий:
Доклиническая стадия. Клинические проявления отсутствуют, однако наблюдается амилоидопатия, что дает экспертной группе NIA-АА основания для выделения данного состояния в качестве стадии БА. Тем не менее, B. Dubois и коллеги считают, что наличие биомаркеров при отсутствии клинических симптомов можно рассматривать как доклиническую стадию БА только при наличии ранних когнитивных или поведенческих нарушений [Н.Н. Коберская, 2017], [Е.И. Гусев, 2018].
Амнестические умеренные когнитивные нарушения (УКН). Больной замечает ухудшения мнестической функции, что подтверждается нейропсихологическими тестами. Другие когнитивные функции не страдают, человек самостоятелен. Деменция отсутствует [Е.И. Гусев, 2018].
Дебют БА (легкая деменция). Для этой стадии характерны малозаметные когнитивные нарушения. Возникают проблемы с памятью: больной не может вспомнить недавние события, значения некоторых слов и с трудом усваивает новую информацию (такой характер регрессии назван законом Рибо). Вероятны также нарушения абстрактного мышления, внимания и способности к планированию поведения. Тем не менее, на ранней стадии больной сохраняет навыки самообслуживания. Критичность мышления сохранна, поэтому человек начинает беспокоиться по поводу своего состояния. В связи с этим возможно развитие тревожно-депрессивных расстройств (депрессия диагностируется у 20-25% таких больных). На МРТ наблюдается атрофия гиппокампа. [Н.Н. Яхно и др., 2011].
Развернутые стадии (умеренная деменция). Если на ранней стадии, больной способен выполнять привычные действия и путешествовать по знакомым маршрутам, то по мере прогрессирования БА, возникают большие трудности в повседневной деятельности. Наблюдается выраженная дезориентировка в пространстве, пространственная апраксия- нарушение движений и агнозия- нарушение восприятия (апракто-агностический синдром). Формируется амнестическая (обеднение словарного запаса в связи с забыванием), а позже и сенсорная (непонимание обращенной и собственной речи) афазии.
Идет сенильная перестройка структуры личности. Человек становится эгоцентричен, агрессивен, ворчлив, начинает безосновательно подозревать близких в злых умыслах (воровство, желание причинить вред больному). Особенности поведения при БА: ухудшение сна, нарушения пищевого поведения, социального взаимодействия (сексуальная несдержанность, утрата тактичности), бесцельная двигательная активность (бродяжничество, перекладывание вещей). [Н.Н. Яхно и др., 2011], [Е.И. Гусев, 2018]
Поздние стадии (тяжелая деменция). Больные полностью утрачивают самостоятельность, становятся беспомощны. Они не могут принимать пищу, справлять нужду, одеваться без помощи посторонних, возникает апраксия ходьбы и одевания. На фоне интеллектуальной недостаточности идет регрессия поведенческих нарушений. Человек становится апатичен, снижаются витальные мотивации. Развивается тотальная афазия (утрата речи как способа коммуникации), возможно формирование мутизма (отказ идти на контакт при сохранной речевой функции). Смерть чаще всего наступает из-за осложнений обездвиженности или интеркуррентных заболеваний (возникших вследствие БА) [Н.Н. Яхно и др., 2011].
Лечение.
Медикаментозное
На данный момент ученые пришли к понимаю того, что терапия БА должна быть комплексной в связи со сложной патофизиологией. Уже начавшееся заболевание невозможно остановить, однако есть способы управлять процессом [P. Colligris et. al., 2018].
Холинергическое направление
Направление основывается на холинергической гипотезе. Ингибиторы ацетилхолинэстеразы позволяют предотвратить недостаток нейромедиатора ацетилхолина. Используются следующие препараты: ривастигмин (экселон), галантамин (реминил), донепезил (арисепт) и ипидакрин (амиридин, аксамон) [М.В. Угрюмов, 2014]. Данная стратегия лечения применяется в основном на стадиях ранней и умеренной деменции, что способствует когнитивному улучшению у 15-40% пациентов с БА [В.В. Захаров и др., 2015], [P. Colligris et. al., 2018].
Глутаматергическое направление
При БА обнаруживается снижение числа глутаматных рецепторов в зоне гиппокампа. Считается, что вследствие данного механизма развивается метаболическая эксайтотоксичность, ведущая к нейродегенерации альцгеймеровского типа [М.В. Угрюмов, 2014]. При глутаматергической терапии на стадиях умеренной и тяжелой деменции используется модулятор глутаматных рецепторов NMDA-типа- мемантин. Длительные исследования показали его эффективность в борьбе с когнитивными нарушениями. Зачастую врачи совмещают холинергическую и глутаматергическую терапию [В.В. Захаров и др., 2015]
Несмотря на то, что данные виды терапии считаются симптоматическими, они могут играть важную роль в патогенетическом воздействии. Так, например, длительное применение донепизила вызывает снижение темпа церебральной атрофии БА, а мемантин защищает постсинаптический нейрон от эксайтотоксического воздействия глутамата и вызванного им постоянного притока кальция в клетку. [В.В. Захаров и др., 2015], [О.С. Левин, Е.Е. Васенина, 2015], [С.И. Гаврилова и др., 2005].
Нейропротективная стратегия
В рамках нейропротективной стратегии применяется Цереброзилин, обладающий нейротрофическими свойствами. Данный препарат снижает отложение βАП, позволяет сохранить синаптические связи за счет замедления фосфорилирования предшественника амилоида. Цереброзилин также положительно влияет на когнитивные функции, снижает выраженность других симптомов БА [М.В. Угрюмов, 2014], [Д. Молчанов, 2007].
Прочие виды терапии
Методы когнитивной интервенции.
Специалисты применяют когнитивное стимулирование (групповые занятия творческими видами деятельности), когнитивный тренинг (выполнение комплекса упражнений), когнитивную реабилитацию (способы компенсации когнитивных нарушений). Эффективность данных методов была подтверждена благодаря нейровизуализации активности значимых для познавательной деятельности отделов коры головного мозга [В.В. Захаров и др., 2015].
Болезнь Паркинсона
БОЛЕЗНЬ ПАРКИНСОНА (БП)- неуклонно прогрессирующее заболевание нервной системы, характеризующееся преимущественной дегенерацией дофаминергических нейронов черного вещества и нарушением функции базальных ганглиев [Н.В. Фёдорова, 2016].
Этиология.
На вопрос причины возникновения БП достоверного ответа ученые еще не нашли. Основными факторами считаются возрастные изменения, генетика и влияние окружающей среды. Их взаимодействие обуславливает риск развития заболевания [Н.В. Фёдорова, 2016].
Внутренние факторы
Возрастные изменения являются ведущим фактором возникновения БП, что связано со снижением пластичности нервной системы [А.А. Таппахов, Т.Я. Николаева, 2016]. В результате изменения в структуре белков не могут быть устранены вовремя, приводя к дегенерации нервных клеток [С.Н. Иллариошкин, 2015].
Генетическая предрасположенность также является одним из наиболее приоритетных факторов, однако семейная форма БП наблюдается лишь в 10% случаев. Данный факт связан с полиэтиологичной природой заболевания. Наличие БП у близких родственников увеличивает риск ее возникновения у примерно в два раза. На данный момент ученые выделили большое количество генов, связанных с развитием БА (табл 1) [А.А. Таппахов, Т.Я. Николаева, 2016].
Таблица 1
Гены и локусы, ассоциированные с болезнью Паркинсона
Локусы |
Гены / белок |
Позиция |
Предполагаемая функция |
Тип наследования |
Начало болезни / фенотип |
Гены и локусы с доказанной ассоциацией с БП |
|||||
P A R K 1 / PARK4 |
SNCA / α-синуклеин |
4q21 |
Пресинаптический белок, компонент телец Леви |
АД |
Раннее / БПРН с быстрым прогрессированием и деменцией |
PARK8 |
LRRK2 / обогащенная повторами лейцина серин-треониновая протеинкиназа 2 |
12q12 |
Протеинкиназа, защита клеток от стрессиндуцированной митохондриальной дисфункции |
АР |
Раннее / БППН, тремор |
PARK2 |
Parkin / убиквитин-Е3- лигаза |
6q25-q27 |
Нейропротективная функция |
АР |
Раннее / ювенильная или БПРН с медленным прогрессированием, дистонией |
PARK6 |
PINK1 / серин, треониновая протеинкиназа |
1p35-p36 |
Митохондриальная киназа, нейропротективная функция |
АР |
Раннее / БПРН с медленным прогрессированием, тремором |
PARK7 |
DJ-1 / белок DJ1 |
1p36 |
Шаперон, антиоксидант |
АР |
Раннее / БПРН, дистония, психотические расстройства |
PARK9 |
ATP13A2 |
1p36 |
Лизосомальная АТФаза |
АР |
Раннее / Ювенильный синдром Куфора-Ракеба, БПРН |
Гены и локусы с возможной ассоциацией с БП |
|||||
PARK3 |
SPR / сепиаптеринре-дуктаза |
2p13 |
Катализация НАДФзависимой редуктазы, участвует в биосинтезе тетрагидробиоп-терина |
АД |
Позднее начало / БППН, деменция |
PARK5 |
UCHL1 / убиквитин-карбоксилконцевая гидролаза L1 |
4p14 |
Убиквитиновая гидролаза |
АД |
Позднее начало / БППН |
PARK10 |
UPS24 / убиквитин-карбоксилконцевая гидролаза 24 |
1p32 |
Участвует в убиквитин-зависимом протеолитическом пути |
Неизвестно |
Позднее начало / БППН |
PARK11 |
GIGYF2 |
2q36-q37 |
Участвует в регуляции рецепторов тирозинкиназы
|
АД |
Позднее начало / БППН |
PARK12 |
Неизвестно |
Xq21-q25 |
Неизвестно |
Неизвестно |
Неизвестно |
PARK13 |
Omi / HTRA2 |
2p13 |
Возможно, участвует в митохондриальной дисфункции |
Неизвестно |
Позднее начало |
PARK16 |
Неизвестно |
1q32 |
Неизвестно |
Неизвестно |
Неизвестно |
|
GBA |
1q21 |
Лизосомальная дегидратация |
Неизвестно |
Вариабельность клиники / деменция / болезнь Гоше |
Примечание: АД – аутосомно-доминантный; АР – аутосомно-рецессивный; БПРН – болезнь Паркинсона с ранним началом; БППН – болезнь Паркинсона с поздним началом.
Внешние факторы
Воздействие внешних факторов способно вызвать процесс нейровоспаления. Конкретными причинами его возникновения могут быть [А.А. Таппахов, Т.Я. Николаева, 2016], [Ю.Н. Васильев, 2013], [Stykel M. G. et al., 2018]:
Инфекционные агенты (в том числе прионы);
Пестициды, гербициды. Провоцируют изменения структуры α-синуклеина, приводящие к гибели нервных клеток;
Тяжелые металлы. Марганец, железо, медь, свинец, алюминий и цинк накапливаются в черном веществе и вызывают окислительный стресс;
МФТП (1-метил-4-фенил-1,2,3,6-тетрагидро-пиридин). Этот протоксин нарушает дыхательный процесс митохондрий и повреждает дофаминергические нейроны черной субстанции;
Черепно-мозговые травмы. Не влияют напрямую, но способны спровоцировать ускорение патологического процесса;
Патогенез.
Основным звеном патогенеза БП считается формирование нейротоксических агрегатов α-синуклеина, возникающие в процессе конформационной перестройки нативной формы белка (растворимой и несвернутой, с неупорядоченной структурой). Выделяют следующие молекулярные механизмы развития БП:
Дисфункция убиквитин-протеасомной системы. Данная система отвечает за регулируемое расщепление белков. Нарушения в ее функционировании вызывают формирование нейротоксических агрегатов α-синуклеина, провоцируя образование телец Леви и последующую гибель нейронов [А.А. Таппахов, Т.Я. Николаева, 2016].
Нарушение регуляции лизосомальной аутофагии. Сбой процесса деградации мутированного α-синуклеина связывают с дегенерацией дофаминергических нейронов [А.А. Таппахов, Т.Я. Николаева, 2016].
Дисфункция митохондрий и окислительный стресс. В результате нарушения клеточного дыхания образуются свободно-радикальные формы кислорода, усугублябющие повреждение митохондрий. Вследствие снижения выработки АТФ возникают эксайтотоксические процессы (гиперактивация NMDA-рецепторов). Эти процессы сопровождаются увеличением притока кальция в клетку, что приводит в конечном итоге к апоптозу [А.А. Таппахов, Т.Я. Николаева, 2016].
Клиническая картина.
При снижении дофамина в хвостатом ядре и скорлупе не менее чем на 70% возникают следующие клинические симптомы [Н.В. Фёдорова, 2016], [Ю.Н. Васильев, 2013]:
Гипокинезия. Снижение двигательной активности- объема движений, их амплитуды, темпа.
Тремор покоя. Непроизвольное сокращение мышц при отсутствии движения. Начинается с верхних конечностей, далее вовлекает ноги и нижнюю челюсть.
Мышечная ригидность (феномен «зубчатого колеса»). Повышенный тонус мышц.
Постуральная неустойчивость. Возникает на более поздних стадиях, характеризуется нарушением ходьбы- частые падения, больной нуждается в дополнительной опоре.
На основе преобладания тех или иных симптомов выделяют несколько клинических форм БП [О. И. Михайлусова, В. А. Куташов, 2015], [Ю.Н. Васильев, 2013]:
Дрожательно-ригидная форма. (Встречается чаще других, главным признаком считается развитие тремора.)
Ригидно-дрожательная форма. (Характеризуется замедленностью движений и повышенным тонусом мышц.)
Акинетико-ригидная форма. (Тремор отсутствует либо проявляется незначительно, например, в периоды волнения.)
Акинетическая форма. (Значимый признак- акинезия, то есть изменение объема произвольных движений.)
Дрожательная форма. (Основным симптомом является дрожание при нормальном или немного повышенном мышечном тонусе, медленно прогрессирует)
Прогрессирование БП отражает шкала Хен-Яра, на основе которой выделяют следующие стадии заболевания:
0.0 стадия — проявления БП отсутствуют;
1.0 стадия — односторонние клинические проявления;
2.0 стадия — симптомы двусторонние без постуральных рефлексов, равновесие не нарушено;
3.0 стадия — клинические проявления умеренные, но двусторонние, постуральная неустойчивость небольшая, пациент не нуждается в посторонней помощи;
4.0 стадия — двигательная активность значительно утрачена, но пациент в состоянии стоять и передвигаться без поддержки;
5.0 стадия — если нет помощи третьего лица, пациент будет прикован к креслу или постели.
Лечение.
Терапия БП основывается на сочетании нескольких факторов воздействия:
Нейропротективная стратегия. Данное направление подразумевает предупреждение развития дегенерационного процесса, восстановление частично поврежденных, но все еще жизнеспособных нейронов и способствование увеличению числа нервных клеток (их имплантация или стимулирование деления имеющихся) [О.С. Левин и др., 2017]
Симптоматическое лечение. Название данной стратегии спорно, так как некоторые препараты способны оказывать патогенетическое воздействие. Лечение направлено на установление нейрохимического баланса в базальных ганглиях. Данный вид терапии основывается на нескольких направлениях [Ю.Н. Васильев, 2013]:
Восполнение дефицита дофамина (L-ДОФА содержащие препараты- мадопар, наком, дуэлин)
Прямая стимуляция рецепторов постсинаптической мембраны нейрона, чувствительных к дофамину (агонисты дофаминовых рецепторов (мирапекс, проноран и 21 реквип модутаб)
Стимуляция высвобождения дофамина из пресинаптического полюса (препараты амантадина -мидантан, симметрел, ПК-Мерц; амфетамин; фенамин.)
Торможение обратного захвата дофамина (трициклические антидепрессанты мелипрамин и амитриптилин; холинолитики)
Снижение катаболизма дофамина и накопление его в синаптической щели. (ингибиторы моноаминоксидазы и катехол-О-метилтрансферазы- селегилин, азилект, сталево, тасмар)
Повышение синтеза дофамина в мозге за счет использования антиоксидантов, предотвращающих ферментные системы от токсического воздействия перекисных радикалов.
Положительное воздействие на тканевой обмен головного мозга (ГАМК, церебролизин, биогенные стимуляторы, витамины, ноотропные и психотропные препараты, антиагреганты)
В зависимости от формы, преобладающих симптомов, стадии заболевания и индивидуальных противопоказаний специалистом подбирается комплексная комбинированная терапия [Ю.Н. Васильев, 2013].
Физическая и социально-психологическая реабилитация. Лечебная физическая культура, речевая гимнастика, нейропсихологический тренинг, психотерапия способствуют улучшению психологического и физического самочувствия больного [Ю.Н. Васильев, 2013].
Функциональная хирургия. Хирургическое вмешательство применяктся в основном на тяжелых стадиях заболевания. Для нормализации функционального состояния проводятся малоинвазивные операции (паллидотомия, таламотомия) и применяются методы хронической электростимуляции подкорковых структур.
Диагностика НДЗ.
Своевременный и точный диагноз в случае НДЗ очень важен, ведь на данный момент не существует способа обратить патологический процесс вспять. Тем не менее, терапия может значительно снизить темпы развития болезни, в связи с чем приоритетом исследований становится изучение ранних биомаркеров [P. Colligris et. al., 2018]. Разработаны специальные диагностические критерии. Например, [Postuma R.B. et al., 2015] при БП, однако их верное использование зависит от компетенции врача. На сегодняшний день момент основными методами диагностики являются:
Компьютерная томография (КТ), магнитно-резонансная томография (МРТ). Довольно дорогие методы, позволяют увидеть анатомическое и функциональное состояние мозга. При нарушениях дают возможность определить очаги поражения. Например, при БА обнаруживается атрофия гиппокампа на коронарных срезах [Р.В. Магжанов, К.З. Бахтиярова, Е.В. Первушина, 2018])
Позитронно-эмиссионная томография (ПЭТ). Позволяет зафиксировать снижение метаболизма и кровотока в медиобазальных отделах лобных долей, глубинных и задних отделах височных долей и в теменных долях головного мозга при БА [Р.В. Магжанов, К.З. Бахтиярова, Е.В. Первушина, 2018]
Анализ крови. Тест «NuroPro», определяя уровень 59 белков–биомаркеров, позволяет установить БА за 6 лет до манифестации с чувствительностью около 90% [А.А. Воробьева, А.В. Васильев, 2009].
Анализ ликвора. Позволяет установить наличие БА за счет определения уровня βАП и тау-протеина в спинномозговой жидкости [А.А. Воробьева, А.В. Васильев, 2009].
Генетический анализ. Выявляет наличие мутаций в генах, которые ассоциируют с НДЗ. Их наличие не всегда определяет развитие заболевания [С.Л. Дудук, 2009]
Тесты на когнитивные функции. «Часы», «Скрытый текст», «Sage-тест».
Для осуществления ранней диагностики необходим комплекс мер: диспансеризация здорового населения, формирование групп риска (на основе выделения биомаркеров) и последующее обследование при помощи более сложных видов диагностики (ПЭТ, МРТ и т.д.). Используемые методы ранней диагностики:
Фармакологическая провокация (Введение центральных холиноликов (скополамина) у пациентов с доклиническими стадиями БА вызывает более выраженные и более продолжительные нарушения памяти, чем у сверстников без биомаркеров амилоидоза) [В.В. Захаров и др., 2015];
Неинвазивное сканирование сетчатки (НДЗ изменения при БА отражаются в структурных и функциональных изменениях нейроретины и сосудистой системы глаза. Согласно недавним исследованиям, с помощью данного метода можно обнаружить амилоидные бляшки и гиперфосфорилированный тау-белок в сетчатке больных БА. На данный момент разрабатываются платформы визуализации сетчатки, которые могут помочь в диагностике заболевания и проверке эффективности терапии) [P. Colligris et. al., 2018];
Анализаторы вокальных биомаркеров (CohMetrix-Dementia) (Использование компьютерных программ, распознающих языковые переменные, характерные для НДЗ, позволяет определить заболевание с чувствительностью 84–90%) [Т.Н. Семенова и др., 2019];
Анализ обонятельной функции (Снижение обоняния появляется на ранних стадиях БА) [В.В. Захаров и др., 2015];
Анализ режима сна (Проблемы со сном возникают задолго до когнитивных нарушений и могут служить их предиктором) [В.Ю. Лобзин и др., 2018].
Возможными признаками для выделения групп риска считаются:
Возраст выше 60-65 лет [С.Н. Иллариошкин, 2015], [В.К. Ахметжанов, Ч.С. Шашкин, Б.Д. Джамантаева, 2016];
Ненормированный режим сна и отдыха [В.Ю. Лобзин, К.А Колмакова, А.Ю. Емелин, 2018], [В.В. Захаров и др., 2015]
Курение способствует развитию атеросклероза, инсульта, что повышает риск возникновения БА. Предполагается, что курение связано с процессом образования амилоидных бляшек [О. С. Левин, Н. А. Трусова, 2013];
Гиподинамия, нарушения пищевого поведения вызывают повышение риска развития БА в связи с возникновением заболеваний сердечно-сосудистой системы, воспалительного процесса, препятствующего клиренсу βА [О. С. Левин, Н. А. Трусова, 2013]
Контакт с пестицидами и гербицидами при БП [Stykel M. G. et al., 2018], [С.Н. Иллариошкин, 2015];
Черепно-мозговые травмы повышают риск БП у носителей специфических полиморфизмов в генах предрасположенности и, в частности, длинного Rep1-промоторного аллеля в гене SNCA [А.А. Таппахов, Т.Я. Николаева, 2016], [С.Н. Иллариошкин, 2015].
Профилактика НДЗ.
Нормализация режима сна и отдыха.
Так, например, клиренс бета-А во время сна возрастает в 10 раз, что позволяет предотвратить патологический процесс БА [В.Ю. Лобзин, К.А Колмакова, А.Ю. Емелин, 2018], [В.В. Захаров и др., 2015]
Регулярная физическая активность.
Рекомендуются пешие прогулки не менее 30 мин три раза в неделю и/или спортивные упражнения. Физическая активность способствует увеличению толщины коры головного мозга, нормализации веса, кровообращения, давления, что, в свою очередь, снижает вероятность развития деменции [В.А. Парфенов, 2014], [А.А. Таппахов, Т.Я. Николаева, 2016].
Оптимизация питания.
Антиоксиданты, находящиеся в овощах и фруктах, снижают патологическое воздействие окислительного стресса, однако данный эффект наблюдается у еще не подверженных заболеванию людей. В целом средиземноморская диета и умеренное потребление вина (250–500 мл в день) ассоциированы с малым риском развития БА [В.А. Парфенов, 2014]. Умеренное употребление кофе способствует предотвращению развития БП у людей, не использующих гормонально-заместительную терапию. Положительное воздействие кофеина может быть объяснено блокадой аденозиновых А2-рецепторов [А.А. Таппахов, Т.Я. Николаева, 2016].
Умственная работа и социальная активность.
Уровень когнитивно-стимулирующей активности в пожилом возрасте связан с риском развития деменции. Патологический процесс, вызванный НДЗ, вызывает гибель большого количества нервных клеток. Один из нейропротективных механизмов ЦНС- наличие большого количества синапсов. Соответственно, когнитивная стимуляция, способствующая возникновению связей между нейронами, играет важную роль в профилактике процесса дегенерации. Этим, помимо большей информированности, можно объяснить низкий риск развития НДЗ у людей с высоким образованием в сравнении с менее образованными. Специалистами также отмечается положительное влияние когнитивного тренинга на поддержание умственных способностей человека. Дефицит когнитивной деятельности, напротив, ассоциирован с более высоким риском развития НДЗ [М.В. Угрюмов, 2014], [В.А. Парфенов, 2014].
Курение.
Предполагается, что в составе сигарет присутствует вещество, которое стимулируя дофаминергические структуры, выполняет нейропротективную функцию при БП [А.А. Таппахов, Т.Я. Николаева, 2016]. Данный эффект связан с носительством определенных аллелей генов GSTP1 и NAT2. Дальнейшие исследования позволят разработать новый подход к терапевтической стратегии лечения БП [С.Н. Иллариошкин, 2015].