

187-2017
.pdfCHAPTER 36 NSAIDs, Antirheumatic Drugs, Nonopioid Analgesics, & Drugs Used in Gout |
653 |
inflammatory cytokines produced by monocytes or macrophages—eg, IL-1, -6, and -12, and TNF-α.
2.Pharmacokinetics: Only 10–20% of orally administered sulfasalazine is absorbed, although a fraction undergoes enterohepatic recirculation into the bowel where it is reduced by intestinal bacteria to liberate sulfapyridine and 5-aminosalicylic acid (see Figure 62–8). Sulfapyridine is well absorbed while 5-aminosalicylic acid remains unabsorbed. Some sulfasalazine is excreted unchanged in the urine whereas sulfapyridine is excreted after hepatic acetylation and hydroxylation. Sulfasalazine’s halflife is 6–17 hours.
3.Indications: Sulfasalazine is effective in RA and reduces radiologic disease progression. It has also been used in juvenile chronic arthritis, PsA, inflammatory bowel disease, AS, and spondyloar- thropathy-associated uveitis. The usual regimen is 2–3 g/d.
4.Adverse Effects: Approximately 30% of patients using sulfasalazine discontinue the drug because of toxicity. Common adverse effects include nausea, vomiting, headache, and rash. Hemolytic anemia and methemoglobinemia also occur, but rarely. Neutropenia occurs in 1–5% of patients, while thrombocytopenia is very rare. Pulmonary toxicity and positive double-stranded DNA (dsDNA) are occasionally seen, but drug-induced lupus is rare. Reversible infertility occurs in men, but sulfasalazine does not affect fertility in women. The drug does not appear to be teratogenic.
TOCILIZUMAB
1.Mechanism of Action: Tocilizumab, a newer biologic humanized antibody, binds to soluble and membrane-bound IL-6 receptors, and inhibits the IL-6-mediated signaling via these receptors. IL-6 is a proinflammatory cytokine produced by different cell types including T cells, B cells, monocytes, fibroblasts, and synovial and endothelial cells. IL-6 is involved in a variety of physiologic processes such as T-cell activation, hepatic acute-phase protein synthesis, and stimulation of the inflammatory processes involved in diseases such as RA and systemic sclerosis (SSc). In a phase 4 superiority study, tocilizumab monotherapy was superior to adalimumab monotherapy for reduction of signs and symptoms of rheumatoid arthritis in patients with incomplete response to MTX.
2.Pharmacokinetics: The half-life of tocilizumab is dosedependent, approximately 11 days for the 4-mg/kg dose and 13 days for the 8-mg/kg dose. IL-6 can suppress several CYP450 isoenzymes; thus, inhibiting IL-6 may restore CYP450 activities to higher levels. This may be clinically relevant for drugs that are CYP450 substrates and have a narrow therapeutic window (eg, cyclosporine or warfarin), and dosage adjustment of these medications may be needed.
Tocilizumab can be used in combination with nonbiologic DMARDs or as monotherapy. In the United States the recommended starting dose for RA is 4 mg/kg intravenously every 4 weeks followed by an increase to 8 mg/kg (not exceeding 800 mg/infusion) dependent on clinical response.
In Europe, the starting dose of tocilizumab is 8 mg/kg up to 800 mg. Tocilizumab dosage in SJIA or PJIA follows an algorithm that accounts for body weight. Additionally, dosage modifications are recommended on the basis of certain laboratory changes such as elevated liver enzymes, neutropenia, and thrombocytopenia.
3.Indications: Tocilizumab is a bDMARD indicated for adult patients with moderately to severely active RA who have had an inadequate response to one or more DMARDs. It is also indicated in patients who are older than 2 years with active SJIA or active PJIA. A recent study showed that it is slightly more effective than adalimumab. There is an ongoing phase 3 study to test its use in SSc.
4.Adverse Effects: Serious infections including tuberculosis, fungal, viral, and other opportunistic infections have occurred. Screening for tuberculosis should be done prior to beginning tocilizumab. The most common adverse reactions are upper respiratory tract infections, headache, hypertension, and elevated liver enzymes.
Neutropenia and reduction in platelet counts occur occasionally, and lipids (eg, cholesterol, triglycerides, LDL, and HDL) should be monitored. GI perforation has been reported when using tocilizumab in patients with diverticulitis and in those using corticosteroids, although it is not clear that this adverse effect is more common than with TNF-α–blocking agents. Demyelinating disorders including multiple sclerosis are rarely associated with tocilizumab use. Fewer than 1% of the patients taking tocilizumab develop anaphylactic reaction. Anti-tocilizumab antibodies develop in 2% of the patients, and these can be associated with hypersensitivity reactions requiring discontinuation.
TNF ` BLOCKING AGENTS
Cytokines play a central rolein the immune response (see Chapter 55) and in RA. Although a wide range of cytokines are expressed in the joints of RA patients, TNF-α appears to be particularly important in the inflammatory process.
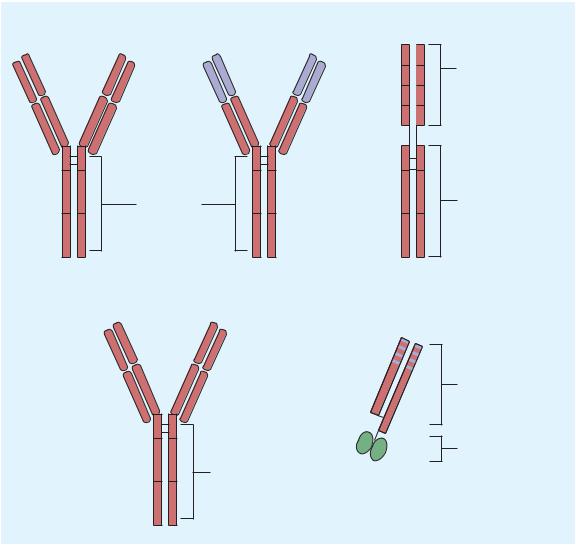
TNF-α affects cellular function via activation of specific mem- brane-bound TNF receptors (TNFR1, TNFR2). Five “legacy” bDMARDs interfering with TNF-α have been approved for the treatment of RA and other rheumatic diseases (Figure 36–4). Biosimilar biologics (bsDMARDs) with lower costs are available in some countries and are being tested in other countries. Thus far, the efficacy, toxicity, and immunogenicity of the biosimilars are equivalent to the legacy compounds. These drugs have many adverse effects in common (see below).
Adalimumab
1.Mechanism of Action: Adalimumab is a fully human IgG1 anti-TNF monoclonal antibody. This compound complexes with soluble TNF-α and prevents its interaction with p55 and p75 cell surface receptors. This results in down-regulation of macrophage and T-cell function.
654 |
SECTION VI Drugs Used to Treat Diseases of the Blood, Inflammation, & Gout |
Adalimumab |
Infliximab |
Etanercept |
VH |
VH |
Extracellular domain |
VL |
VL |
of human p75 receptor |
CH1 |
CH1 |
|
CL |
CL |
|
CH2 |
CH2 |
CH2 |
FC region of |
|
FC region of |
human IgG1 |
|
human IgG1 |
CH3 |
CH3 |
CH3 |
Golimumab |
Certolizumab |
VH |
|
VL |
|
CH1 |
Humanized Fab |
CL |
fragment |
CH2 |
Polyethylene |
|
glycol (PEG) |
|
FC region of |
CH3 |
human IgG1 |
|
FIGURE 36 4 Structures of TNF-α antagonists used in rheumatoid arthritis. CH, constant heavy chain; CL, constant light chain; Fc, complex immunoglobulin region; VH, variable heavy chain; VL, variable light chain. Red regions, human derived; blue regions, mouse derived; green regions, polyethylene glycol (PEG).
2.Pharmacokinetics: Adalimumab is given subcutaneously and has a half-life of 10–20 days. Its clearance is decreased by more than 40% in the presence of methotrexate, and the formation of human anti-monoclonal antibody is decreased when methotrexate is given at the same time. The usual dose in RA is 40 mg every other week, but dosing is frequently increased to 40 mg weekly. In psoriasis, 80 mg is given at week 0, 40 mg at week
1, and then 40 mg every other week thereafter. The initial dose in inflammatory bowel disease is higher; patients receive 160 mg at week 0, and 80 mg 2 weeks later, followed by a 40-mg maintenance dose every other week. Patients with ulcerative colitis should continue maintenance treatment beyond 8 weeks if they show evidence of remission by that time. Adalimumab dose depends on the body weight in patients with JIA: 20 mg every other week for patients weighing 15–30 kg, and 40 mg every other week in patients weighing 30 kg or more.
3.Indications: The compound is approved for RA, AS, PsA, JIA, plaque psoriasis, Crohn’s disease, and ulcerative colitis. It decreases the rate of formation of new erosions. It is effective both as monotherapy and in combination with methotrexate and other nonbiologic csDMARDs. Based only on case reports and case series, adalimumab has also been found to be effective in the treatment of Behçet’s disease, sarcoidosis, and notably, noninfectious uveitis.
Certolizumab
1.Mechanism of Action: Certolizumab is a recombinant, humanized antibody Fab fragment conjugated to a polyethylene glycol (PEG) with specificity for human TNF-α. Certolizumab neutralizes membrane-bound and soluble TNF-α in a dose-dependent manner. Additionally, certolizumab does not
CHAPTER 36 NSAIDs, Antirheumatic Drugs, Nonopioid Analgesics, & Drugs Used in Gout |
655 |
contain an Fc region, found on a complete antibody, and does not fix complement or cause antibody-dependent cell-mediated cytotoxicity in vitro.
2.Pharmacokinetics: Certolizumab is given subcutaneously and has a half-life of 14 days. Methotrexate decreases the appearance of anti-certolizumab antibodies. The usual dose for RA is 400 mg initially and at weeks 2 and 4, followed by 200 mg every other week, or 400 mg every 4 weeks.
3.Indications: Certolizumab is indicated for the treatment of adults with moderately to severely active RA. It can be used as monotherapy or in combination with nonbiologic DMARDs. Additionally, certolizumab is approved in adult patients with
Crohn’s disease, active PsA, and active AS. The certolizumab head-to-head TNFi trial, Exxelerate (NCT01500278), was a multicenter, single-blind, 24-month, randomized, parallelgroup trial in moderate to severe MTX-incomplete-responder RA patients, comparing adalimumab + MTX to certolizumab
+MTX. ACR20 responses at 3 months and achievement of low disease activity at 2 years were numerically comparable for both protocols. Although putatively a 24-month trial, patients could switch from one regimen to the other at 3 months, confounding comparability beyond that time frame. Not surprisingly, given this confounding, the primary goal of certolizumab
+MTX superiority was not met. Patients were switched without washout so blood levels of TNFis as a group could be expected to be very high during the switchover. Interestingly, no serious infectious events occurred during the switch-over period.
Etanercept
1.Mechanism of Action: Etanercept is a recombinant fusion protein consisting of two soluble TNF p75 receptor moieties
linked to the Fc portion of human IgG1 (Figure 36–4); it binds TNF-α molecules and also inhibits lymphotoxin α.
2.Pharmacokinetics: Etanercept is given subcutaneously as 25 mg twice weekly or 50 mg weekly. In psoriasis, 50 mg is given twice weekly for 12 weeks and then is followed by 50 mg weekly. The drug is slowly absorbed, with peak concentration 72 hours after drug administration. Etanercept has a mean serum elimination half-life of 4.5 days. A recent study demonstrated a reduction of radiographic progression with the use of 50 mg of etanercept weekly.
3.Indications: Etanercept is approved for the treatment of RA, juvenile chronic arthritis, psoriasis, PsA, and AS. It can be used as monotherapy, although over 70% of patients taking etanercept are also using methotrexate. Etanercept decreases the rate of formation of new erosions relative to methotrexate alone. It is also being used in other rheumatic syndromes such as scleroderma, granulomatosis with polyangiitis (Wegener’s granulomatosis), giant cell arteritis, Behçet’s disease, uveitis, and sarcoidosis. However, a comparative study of ustekinumab (an IL-12 and IL-23 blocker) and etanercept concluded that ustekinumab at a dose of 45 or 90 mg was superior to highdose etanercept (50 mg twice weekly) over a 12-week period in patients with psoriasis.
Golimumab
1.Mechanism of Action: Golimumab is a human monoclonal
antibody with a high affinity for soluble and membrane-bound TNF-α. Golimumab effectively neutralizes the inflammatory effects produced by TNF-α seen in diseases such as RA.
2.Pharmacokinetics: Golimumab is administered subcutaneously and has a half-life of approximately 14 days. Concomitant use with methotrexate increases golimumab serum levels and decreases anti-golimumab antibodies. The recommended dose for the treatment of RA, PsA, and AS is 50 mg given every
4 weeks. A higher dose of golimumab is used for the treatment of ulcerative colitis as follows: 200 mg initially at week 0 followed by 100 mg at week 2 and every 4 weeks thereafter.
3.Indications: Golimumab with methotrexate is indicated for the treatment of moderately to severely active RA in adult patients. It is also indicated for the treatment of PsA and AS and moderate to severe ulcerative colitis.
Infliximab
1.Mechanism of Action: Infliximab (Figure 36–4) is a chimeric
(25% mouse, 75% human) IgG1 monoclonal antibody that binds with high affinity to soluble and possibly membranebound TNF-α. Its mechanism of action probably is the same as that of adalimumab.
2.Pharmacokinetics: Infliximab is given as an intravenous infusion with “induction” at 0, 2, and 6 weeks and maintenance every 8 weeks thereafter. Dosing is 3–10 mg/kg, and the usual dose is 3–5 mg/kg every 8 weeks. There is a relationship between serum concentration and effect, although individual clearances vary markedly. The terminal half-life is 9–12 days without accumulation after repeated dosing at the recommended interval of 8 weeks. After intermittent therapy, infliximab elicits human antichimeric antibodies in up to 62% of patients. Concurrent therapy with methotrexate markedly decreases the prevalence of human antichimeric antibodies.
3.Indications: Infliximab is approved for use in RA, AS, PsA, Crohn’s disease, ulcerative colitis, pediatric inflammatory bowel disease, and psoriasis. It is being used off-label in other diseases, including granulomatosis with polyangiitis (Wegener’s granulomatosis), giant cell arteritis, Behçet’s disease, uveitis, and sarcoidosis. In RA, infliximab plus methotrexate decreases the rate of formation of new erosions. Although it is recommended that methotrexate be used in conjunction with infliximab, a number of other nonbiologic csDMARDs, including antimalarials, azathioprine, leflunomide, and cyclosporine, can be used as background therapy for this drug. Infliximab is also used as monotherapy.
Adverse Effects ofTNF-`-Blocking Agents
TNF-α–blocking agents have multiple adverse effects in common. The risk of bacterial infections and macrophage-dependent infection (including tuberculosis, fungal, and other opportunistic

656 |
SECTION VI Drugs Used to Treat Diseases of the Blood, Inflammation, & Gout |
infections) is increased, although it remains very low. Activation of latent tuberculosis is lower with etanercept than with other TNF-α–blocking agents. Nevertheless, all patients should be screened for latent or active tuberculosis before starting TNF-α– blocking agents. The use of TNF-α–blocking agents is also associated with increased risk of HBV reactivation; screening for HBV is important before starting the treatment.
TNF-α–blocking agents increase the risk of skin cancers— including melanoma—which necessitates periodic skin examination, especially in high-risk patients. On the other hand, there is no clear evidence of increased risk of solid malignancies or lymphomas with TNF-α–blocking agents, and their incidence may not be different compared with other bDMARDs or active RA itself.
A low incidence of newly formed dsDNA antibodies and antinuclear antibodies (ANAs) has been documented when using TNF-α–blocking agents, but clinical lupus is extremely rare and the presence of ANA and dsDNA antibodies per se does not contraindicate the use of TNF-α–blocking agents. In patients with borderline or overt heart failure (HF), TNF-α–blocking agents can exacerbate HF. TNF-α–blocking agents can induce the immune system to develop antidrug antibodies in about 17% of cases. These antibodies may interfere with drug efficacy and correlate with infusion site reactions. Injection site reactions occur in 20–40% of patients, although they rarely result in discontinuation of therapy. Cases of alopecia areata, hypertrichosis, and erosive lichen planus have been reported. Cutaneous pseudo-lymphomas are reported rarely with TNF-α–blocking agents, especially infliximab. TNF-α–blocking agents may increase the risk of gastrointestinal ulcers and large bowel perforation including diverticular and appendiceal perforation.
Nonspecific interstitial pneumonia, psoriasis, and sarcoidosislike syndrome are among the rare reported toxicities associated with TNF-α blockers. Rare cases of leukopenia, neutropenia, thrombocytopenia, and pancytopenia have also been reported. The precipitating drug should be discontinued in such cases.
USTEKINUMAB
1.Mechanism of Action: Ustekinumab is an IL-12 and IL-23 antagonist. It is a fully human IgG monoclonal antibody to the p40 protein subunit, which is part of both IL-12 and IL-23. These two cytokines are important contributors to the chronic inflammation in psoriasis plaques, PsA, and Crohn’s disease. Ustekinumab prevents the binding of the p40 subunit of both IL-12 and IL-23 to the IL-12 receptor b1 found on the surface of CD4 T cells and NK cells. This interruption interferes with IL-12 and IL-23 signal transduction and suppresses the formation of proinflammatory TH1 and TH17 cells.
2.Pharmacokinetics: Ustekinumab is available as a 45and 90-mg SC injection for PsA and plaque psoriasis. Its bioavailability is 57% following SC injection; time to peak plasma concentration is 7–13.5 days and elimination half-life is 10–126 days. For adults with PsA, a loading dose at 0 and 4 weeks is followed by maintenance doses once every 12 weeks. IV infusion as a 130 mg dose is available for Crohn’s disease.
3.Indications: Ustekinumab is indicated for treatment of adult patients with PsA. It can be used as monotherapy or in combination with methotrexate. Other indications include plaque psoriasis and Crohn’s disease.
4.Adverse Effects: Upper respiratory tract infection is the most common side effect, but rare severe infection, malignancy, and reversible posterior leukoencephalopathy syndrome have been reported. Ustekinumab should be discontinued at least 15 weeks before live vaccines are administered and can be resumed at least 2 weeks after.
SECUKINUMAB
1.Mechanism of Action: Secukinumab is a human IgG1 monoclonal antibody that selectively binds to the IL-17A cytokine, inhibiting its interaction with the IL-17A receptor. IL-17A is involved in normal inflammatory and immune responses. Elevated concentrations of IL-17A are found in psoriatic plaques and PsA.
2.Pharmakokinetics: Secukinumab is available as a SC injection or lyophilized powder for injection. Its peak plasma concentration is 13.7 mcg/mL (150 mg dose) and 27.3 mcg/mL (300 mg dose); elimination half-life is 22–31 days.
3.Indications and Dosage: Secukinumab is indicated for moderate to severe plaque psoriasis in patients who are candidates for systemic therapy or phototherapy. Initial loading dose is 300 mg SC at weeks 0, 1, 2, 3, and 4, followed by monthly maintenance (300 mg SC or 150 mg SC monthly). For adults with active PsA and moderate to severe plaque psoriasis, the same recommendations are followed. For patients with psoriatic arthritis as well as AS, administer with or without a loading dosage by SC injection; 150 mg SC every 4 weeks with or without MTX is recommended.
4.Adverse Effects: As for any of these biologics, infection is a common side effect (28.7%). Nasopharyngitis occurs in about 12%. TB status should be evaluated prior to therapy. Secukinumab may exacerbate Crohn’s disease.
TOFACITINIB
1.Mechanism of Action: Tofacitinib is a targeted synthetic small molecule (tsDMARD) that selectively inhibits all members of the Janus kinase (JAK; see Chapter 2) family to varying degrees. At therapeutic doses, tofacitinib exerts its effect mainly by inhibiting JAK3, and to a lesser extent JAK1, hence interrupting the JAK-STAT signaling pathway. This pathway plays a major role in the pathogenesis of autoimmune diseases including RA. The JAK3/JAK1 complex is responsible for signal transduction from the common γ-chain receptor (IL2RG) for IL-2, -4, -7, -9, -15, and -21, which subsequently influences transcription of several genes that are crucial for the differentiation, proliferation, and function of NK cells and T and B lymphocytes. In addition, JAK1 (in combination with

CHAPTER 36 NSAIDs, Antirheumatic Drugs, Nonopioid Analgesics, & Drugs Used in Gout |
657 |
other JAKs) controls signal transduction from IL-6 and interferon receptors. RA patients receiving tofacitinib rapidly reduce C-reactive protein.
2.Pharmacokinetics: The recommended dose of tofacitinib in the treatment of RA is 5 mg twice daily; there is a clear trend to increased response (and increased toxicity) at double this dose. In 2016, the FDA approved extended-release (XR) tofacitinib citrate 11 mg tablets for once-daily treatment. Tofacitinib has an absolute oral bioavailability of 74%, highfat meals do not affect the AUC, and the elimination half-life is about 3 hours. Metabolism (of 70%) occurs in the liver, mainly by CYP3A4 and to a lesser extent by CYP2C19. The remaining 30% is excreted unchanged by the kidneys. Patients taking CYP enzyme inhibitors and those with moderate hepatic or renal impairment require dose reduction to 5 mg once daily. It should not be given to patients with severe hepatic disease.
3.Indications: Tofacitinib was originally developed to prevent solid organ allograft rejection. It has also been tested for the treatment of inflammatory bowel disease, spondyloarthritis, psoriasis, and dry eyes. To date, tofacitinib is approved in the United States for the treatment of adult patients with moderately to severely active RA who have failed or are intolerant to methotrexate. It is not approved in Europe for this indication. It can be used as a monotherapy or in combination with other csDMARDs, including methotrexate. Ongoing studies are evaluating its role in other rheumatic diseases such as PsA, psoriasis, and JIA.
4.Adverse Effects: Tofacitinib slightly increases the risk of infection, and it has thus far not been used with potent immunosuppressants (eg, azathioprine, cyclosporine) or biologic bDMARDs because additive immunosuppression is feared, although it has not been tested in combinations. Upper respiratory tract infection and urinary tract infection represent the most common infections. More serious infections are also reported, including pneumonia, cellulitis, esophageal candidiasis, and other opportunistic infections. All patients should be screened for latent or active tuberculosis before the initiation of treatment. Lymphoma and other malignancies such as lung and breast cancer have been reported in patients taking tofacitinib, although some studies discuss the potential use of JAK inhibitors to treat certain lymphomas. Dose-dependent increases in the levels of low-density lipoprotein (LDL), high-density lipoprotein (HDL), and total cholesterol have been found in patients receiving tofacitinib, often beginning about 6 weeks after starting treatment; therefore, lipid levels should be monitored. Although tofacitinib causes a dose-dependent increase in CD19 B cells and CD4 T cells plus a reduction in CD16/CD56 NK cells, the clinical significance of these changes remains unclear. Drug-related neutropenia and anemia occur, requiring drug discontinuation. Headache, diarrhea, elevation of liver enzymes, and gastrointestinal perforation are among the other reported effects of tofacitinib.
INTERLEUKIN 1 INHIBITORS
IL-1α plays a major role in the pathogenesis of several inflammatory and autoimmune diseases including RA. IL-1α, IL-1β, and IL-1 receptor antagonist (IL-1RA) are other members of the IL-1 family. All three bind to IL-1 receptors in the same manner. However, IL1RA does not initiate the intracellular signaling pathway and thus acts as a competitive inhibitor of the proinflammatory IL-1α and IL-1β.
Anakinra
1.Mechanism of Action: Anakinra is the oldest drug in this family but is now rarely used for RA.
2.Pharmacokinetics: Anakinra is administered subcutaneously and reaches a maximum plasma concentration after 3–7 hours. The absolute bioavailability of anakinra is 95%, and it has a 4- to 6-hour terminal half-life. The recommended dose in the treatment of RA is 100 mg daily. The dose of anakinra depends on the body weight in the treatment of cryopyrin-associated periodic syndrome (CAPS), starting with 1–2 mg/kg per day to a maximum of 8 mg/kg per day. Reduction in the frequency of administering anakinra to every other day is recommended in patients with renal insufficiency.
3.Indications: Anakinra is approved for the treatment of moderately to severely active RA in adult patients, but it is rarely used for this indication. However, anakinra is the drug of choice for CAPS, particularly the neonatal-onset multisystem inflammatory disease (NOMID) subtype. Anakinra is effective in gout (see below) and is used for other diseases including Behçet’s disease and adult onset JIA. Its use for giant cell arteritis is controversial.
Canakinumab
1.Mechanism of Action: Canakinumab is a human IgG1/κ monoclonal antibody against IL-1β. It forms a complex with IL-1β, preventing its binding to IL-1 receptors.
2.Pharmacokinetics: Canakinumab is given by subcutaneous injection. It reaches peak serum concentrations 7 days after a single subcutaneous injection. Canakinumab has an absolute bioavailability of 66% and a 26-day mean terminal half-life. The recommended dose for patients with SJIA who weigh more than 7.5 kg is 4 mg/kg every 4 weeks. There is a weightadjusted algorithm for treating CAPS.
3.Indications: Canakinumab is indicated for active SJIA in children 2 years or older. As noted, it is also used to treat CAPS, particularly the familial cold autoinflammatory syndrome and Muckle-Wells syndrome subtypes for adults and children 4 years or older. Canakinumab is also used to treat gout (see below).
Rilonacept
1.Mechanism of Action: Rilonacept is the ligand-binding domain of the IL-1 receptor. It binds mainly to IL-1β and binds with lower affinity to IL-1α and IL-1RA. Rilonacept neutralizes IL-1β and prevents its attachment to IL-1 receptors.

658 |
SECTION VI Drugs Used to Treat Diseases of the Blood, Inflammation, & Gout |
2. Pharmacokinetics: The subcutaneous dose of rilonacept for CAPS is age-dependent. In patients 12–17 years of age, 4.4 mg/kg (maximum of 320 mg) is the loading dose, with a maintenance dose of 2.2 mg/kg (maximum of 160 mg) weekly. Those 18 years and older receive 320 mg as a loading dose and 160 mg weekly thereafter. The steady-state plasma concentration is reached after 6 weeks.
3.Indications: Rilonacept is approved to treat CAPS subtypes: familial cold autoinflammatory syndrome and Muckle-Wells syndrome in patients 12 years or older. Rilonacept is also used to treat gout (see below).
Adverse Effects of Interleukin-1 Inhibitors
The most common adverse effects are injection site reactions (up to 40%) and upper respiratory tract infections. Serious infections occur rarely in patients given IL-1 inhibitors. Headache, abdominal pain, nausea, diarrhea, arthralgia, and flu-like illness all have been reported, as have hypersensitivity reactions. Patients taking IL-1 inhibitors may experience transient neutropenia, which requires regular monitoring of neutrophil counts.
the basis of complementary mechanisms of action, nonoverlapping pharmacokinetics, and nonoverlapping toxicities.
When added to methotrexate background therapy, cyclosporine, chloroquine, hydroxychloroquine, leflunomide, infliximab, adalimumab, rituximab, and etanercept have all shown improved efficacy. Triple therapy with methotrexate, sulfasalazine, and hydroxychloroquine appears to be as effective as etanercept and methotrexate. In contrast, azathioprine or sulfasalazine plus methotrexate results in no additional therapeutic benefit. Other combinations have occasionally been used.
While it might be anticipated that combination therapy could result in more toxicity, this is often not the case. Combination therapy for patients not responding adequately to monotherapy is now the rule in the treatment of RA.
GLUCOCORTICOID DRUGS
The general pharmacology of corticosteroids, including mechanism of action, pharmacokinetics, and other applications, is discussed in Chapter 39.
BELIMUMAB
Belimumab is an antibody that specifically inhibits B-lymphocyte stimulator (BLyS). It is administered as an intravenous infusion. The recommended dose is 10 mg/kg at weeks 0, 2, and 4, and every 4 weeks thereafter. Belimumab has a distribution half-life of 1.75 days and a terminal half-life of 19.4 days.
Belimumab is approved only for the treatment of adult patients with active, seropositive SLE who are receiving standard treatment. The drug was approved after a protracted series of clinical trials, and its place in the SLE armamentarium is not clear. Belimumab should not be used in patients with active renal or neurological manifestations of SLE, as there are no data for these conditions. In addition, the efficacy of belimumab has not been tested in combination with other bDMARDs or cyclophosphamide.
The most common adverse effects of belimumab are nausea, diarrhea, and respiratory tract infection. As with other bDMARDs, there is a slight increase in the risk of infection including serious infections. Cases of depression and suicide have been reported in patients receiving belimumab, although these patients may have had neurologic SLE, thus confounding the causal relationship. Infusion reactions including anaphylaxis are among the other adverse effects. A very small percentage of patients develop antibodies toward belimumab; their clinical significance is not clear.
COMBINATION THERAPY WITH
DMARDs
In a 1998 survey, approximately half of North American rheumatologists treated moderately aggressive RA with combination therapy, and the use of drug combinations is probably much higher now. Combinations of DMARDs can be designed rationally on
Indications
Corticosteroids have been used in 60–70% of RA patients. Their effects are prompt and dramatic, and they are capable of slowing the appearance of new bone erosions. Corticosteroids may be administered for certain serious extra-articular manifestations of RA such as pericarditis or eye involvement or during periods of exacerbation. When prednisone is required for long-term therapy, the dosage should not exceed 7.5 mg daily, and gradual reduction of the dose should be encouraged. Alternate-day corticosteroid therapy is usually unsuccessful in RA.
Other rheumatic diseases in which the corticosteroids’ potent anti-inflammatory effects may be useful include vasculitis, SLE, Wegener’s granulomatosis, PA, giant cell arteritis, sarcoidosis, and gout. Intra-articular corticosteroids are often helpful to alleviate painful symptoms and, when successful, are preferable to increasing the dosage of systemic medication.
Some of the symptoms of RA, especially morning stiffness and joint pain, follow a circadian rhythm, probably due to an increase in proinflammatory cytokines in the early morning. A recent approach uses delayed-release prednisone for the treatment of early morning stiffness and pain in RA. The tablet contains an inactive outer layer and a core of the active drug. The outer layer dissolves over 4–6 hours, releasing the prednisone. Taking the drug at 9–10 pm results in a small pulse of prednisone at 2–4 am, decreasing the circadian inflammatory cytokines. At low doses of 3–5 mg prednisone, the adrenal-pituitary axis does not seem to be impacted.
Adverse Effects
Prolonged use of corticosteroids leads to serious and disabling toxic effects as described in Chapter 39. Many of these adverse effects occur at doses below 7.5 mg prednisone equivalent daily and many experts believe that even 3–5 mg/d can cause adverse

CHAPTER 36 NSAIDs, Antirheumatic Drugs, Nonopioid Analgesics, & Drugs Used in Gout |
659 |
effects in susceptible individuals when this class of drugs is used over prolonged periods.
■ OTHER ANALGESICS
Acetaminophen is one of the most important drugs used in the treatment of mild to moderate pain when an anti-inflammatory effect is not necessary. Phenacetin, a prodrug that is metabolized to acetaminophen, is more toxic and should not be used.
ACETAMINOPHEN
Acetaminophen is the active metabolite of phenacetin and is responsible for its analgesic effect. It is a weak COX-1 and COX-2 inhibitor in peripheral tissues and possesses no significant antiinflammatory effects.
|
|
|
|
|
|
H |
|
O |
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
HO |
|
|
|
|
|
N |
|
C |
|
CH3 |
|||
|
|
|
|
||||||||||
4 g/d are not usually recommended, and a history of alcoholism contraindicates even this dose. Early symptoms of hepatic damage include nausea, vomiting, diarrhea, and abdominal pain. Cases of renal damage without hepatic damage have occurred, even after usual doses of acetaminophen. Therapy for overdose is much less satisfactory than that for aspirin overdose. In addition to supportive therapy, one should provide sulfhydryl groups in the form of acetylcysteine to neutralize the toxic metabolites (see Chapter 58).
5.Hemolytic anemia and methemoglobinemia are very rare adverse events. Interstitial nephritis and papillary necrosis— serious complications of phenacetin—have not occurred, and GI bleeding also has not occurred. Caution is necessary in patients with any type of liver disease.
6.Dosage: Acute pain and fever may be effectively treated with 325–500 mg four times daily and proportionately less for children. Dosing in adults is now recommended not to exceed 4 g/d, in most cases.
KETOROLAC
1.Pharmacokinetics: Acetaminophen is administered orally. Peakbloodconcentrationsareusuallyreachedin30–60 minutes. Acetaminophen is poorly bound to plasma proteins and is partially metabolized by hepatic microsomal enzymes to the inactive sulfate and glucuronide (see Figure 4–5). Less than 5% is excreted unchanged. In large doses, a minor but highly reactive metabolite (N-acetyl-p-benzoquinone) is important because it is toxic to both liver and kidney (see Chapter 4). The half-life of acetaminophen is 2–3 hours and is relatively unaffected by renal function. With toxic doses or liver disease, the half-life may be increased twofold or more.
2.Indications: Although said to be equivalent to aspirin as an analgesic and antipyretic agent, acetaminophen lacks antiinflammatory properties. It does not affect uric acid levels and lacks platelet-inhibiting effects. The drug is useful in mild to moderate pain such as headache, myalgia, postpartum pain, and other circumstances in which aspirin is an effective analgesic. Acetaminophen alone is inadequate therapy for inflammatory conditions such as RA. For mild analgesia, acetaminophen is the preferred drug in patients allergic to aspirin, when salicylates are poorly tolerated. It is preferable to aspirin in patients with hemophilia, in those with a history of peptic ulcer, and in those in whom bronchospasm is precipitated by aspirin. Unlike aspirin, acetaminophen does not antagonize the effects of uricosuric agents.
3.Adverse Effects: In therapeutic doses, a mild reversible increase in hepatic enzymes may occasionally occur. With larger doses, dizziness, excitement, and disorientation may occur. Ingestion of 15 g of acetaminophen may be fatal, death being caused by severe hepatotoxicity with centrilobular necrosis, sometimes associated with acute renal tubular necrosis (see Chapters 4 and 58).
4.Present data indicate that even 4 g acetaminophen is associated withincreasedliverfunctiontestabnormalities.Dosesgreaterthan
Ketorolac is an NSAID promoted for systemic use mainly as a short-term analgesic (not longer than 1 week), not as an antiinflammatory drug (although it has typical NSAID properties). Pharmacokinetics are presented in Table 36–1. The drug is an effective analgesic and has been used successfully to replace morphine in some situations involving mild to moderate postsurgical pain. It is most often given intramuscularly or intravenously, but an oral formulation is available. When used with an opioid, it may decrease the opioid requirement by 25–50%. Toxicities are similar to those of other NSAIDs (see page 645), although renal toxicity is more common with chronic use.
TRAMADOL
Tramadol is a centrally acting synthetic analgesic, structurally related to opioids. Since naloxone, an opioid receptor blocker, inhibits only 30% of the analgesic effect of tramadol, the mechanism of action of this drug must involve both nonopioid and opioid receptors. Tramadol does not have significant anti-inflammatory effects. The drug may exert part of its analgesic effect by enhancing 5-hydroxytryptamine (5-HT) release and inhibiting the reuptake of norepinephrine and 5-HT (see Chapter 31).
■ DRUGS USED IN GOUT
Gout is a metabolic disease characterized by recurrent episodes of acute arthritis due to deposits of monosodium urate in joints and cartilage. Uric acid renal calculi, tophi, and interstitial nephritis may also occur. Adverse cardiovascular outcomes are becoming more clear as well. Gout is usually associated with a high serum uric acid level (hyperuricemia), a poorly soluble substance that is the major end product of purine metabolism. In most mammals,

660 |
SECTION VI Drugs Used to Treat Diseases of the Blood, Inflammation, & Gout |
uricase converts uric acid to the more soluble allantoin; this enzyme is absent in humans. While clinical gouty episodes are associated with hyperuricemia, most individuals with hyperuricemia may never develop a clinical event from urate crystal deposition.
The treatment of gout aims to relieve acute gouty attacks and prevent recurrent gouty episodes and urate lithiasis. Therapies for acute gout are based on our current understanding of the pathophysiologic events that occur in this disease (Figure 36–5). Clinical gout is dependent on a macromolecular complex of proteins, called NLRP3, which regulates the activation of IL-1. Urate crystals activate NLRP3, resulting in release of prostaglandins and lysosomal enzymes by synoviocytes. Attracted by these chemotactic mediators, polymorphonuclear leukocytes migrate into the joint space and amplify the ongoing inflammatory process. In the later phases of the attack, increased numbers of mononuclear phagocytes (macrophages) appear, ingest the urate crystals, and release more inflammatory mediators.
Before starting chronic urate-lowering therapy for gout, patients in whom hyperuricemia is associated with gout and urate lithiasis must be clearly distinguished from individuals with only hyperuricemia. The efficacy of long-term drug treatment in an asymptomatic hyperuricemic person is unproved. Although there are data suggesting a clear relationship between the degree of uric acid elevation and the likelihood of clinical gout, in some individuals, uric acid levels may be elevated up to 2 standard deviations above the mean for a lifetime without adverse consequences. Many different agents have been used for the treatment of acute and chronic gout. However, non-adherence to these drugs is exceedingly common; adherence has been documented to be 18–26% in younger patients. Providers should be aware of compliance as an important issue.
Synoviocytes |
|
|
|
Colchicine |
|
||
Urate |
– |
LTB4 |
|
crystal |
|
|
|
PMN |
|
|
|
PG |
|
PG |
|
Enzymes IL-1 |
|
|
|
|
|
PG |
|
|
|
MNP |
– |
|
|
|
– |
IL-1 |
|
|
Indomethacin, |
|
|
|
|
|
|
|
phenylbutazone |
FIGURE 36 5 Pathophysiologic events in a gouty joint. Synoviocytes phagocytose urate crystals and then secrete inflammatory mediators, which attract and activate polymorphonuclear leukocytes (PMN) and mononuclear phagocytes (MNP) (macrophages). Drugs active in gout inhibit crystal phagocytosis and polymorphonuclear leukocyte and macrophage release of inflammatory mediators. PG, prostaglandin; IL-1, interleukin-1; LTB4, leukotriene B4.
COLCHICINE
Although NSAIDs, corticosteroids, or colchicine are now first-line drugs for acute gout, colchicine was the primary treatment for many years. Colchicine is an alkaloid isolated from the autumn crocus, Colchicum autumnale. Its structure is shown in Figure 36–6.
1.Pharmacokinetics: Colchicine is absorbed readily after oral administration, reaches peak plasma levels within 2 hours, and is eliminated with a serum half-life of 9 hours. Metabolites are excreted in the intestinal tract and urine.
2.Pharmacodynamics: Colchicine relieves the pain and inflammation of gouty arthritis in 12–24 hours without altering the metabolism or excretion of urates and without other analgesic effects. Colchicine produces its anti-inflammatory effects by binding to the intracellular protein tubulin, thereby preventing its polymerization into microtubules and leading to the inhibi-
tion of leukocyte migration and phagocytosis. It also inhibits
the formation of leukotriene B4 and IL-1β. Several of colchicine’s adverse effects are produced by its inhibition of tubulin polymerization and cell mitosis.
3.Indications: Colchicine is indicated for gout and is also used between attacks (the “intercritical period”) for prolonged
H3C |
|
|
|
O |
|
|
|
|
|
|
|
|
|
|
|
|
H |
|
O |
|
|
||||||
|
|
|
|
|
|
|
|
||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
H3C |
|
|
|
O |
|
|
|
|
|
|
|
|
|
|
|
|
N |
|
C |
|
CH3 |
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CH3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O |
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O |
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CH3 |
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
Colchicine |
|
|
|
|
|
|
|||||||||||||
H3C |
|
|
CH2 |
|
CH2 |
|
O |
|
|
|
|
O |
|
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
|
|
|
|
||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
H3C |
|
|
|
CH2 |
|
|
N |
|
S |
|
|
|
|
C |
|
|
OH |
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
|
|
|
CH2 |
|
|
|
|
|
|
|
|
|
|
||||||||||||||
|
O |
|
|
|
|
|
|
||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||
Probenecid
|
|
|
|
|
|
O |
N |
|
|
|
|
|
|
|
N |
||
O |
||||||||
|
||||||||
|
|
|
|
|
|
|
|
|
S |
|
CH2 |
|
CH2 |
O |
|||
|
|
|||||||
Sulfinpyrazone
FIGURE 36 6 Colchicine and uricosuric drugs.

CHAPTER 36 NSAIDs, Antirheumatic Drugs, Nonopioid Analgesics, & Drugs Used in Gout |
661 |
prophylaxis (at low doses). It prevents attacks of acute Mediterranean fever and may have a mild beneficial effect in sarcoid arthritis and in hepatic cirrhosis. Colchicine is also used to treat and prevent pericarditis, pleurisy, and coronary artery disease, probably due to its anti-inflammatory effect. Although it has been given intravenously, this route is no longer approved by the FDA (2009).
4.Adverse Effects: Colchicine often causes diarrhea and may occasionally cause nausea, vomiting, and abdominal pain. Hepatic necrosis, acute renal failure, disseminated intravascular coagulation, and seizures have also been observed. Colchicine may rarely cause hair loss and bone marrow depression, as well as peripheral neuritis, myopathy, and, in some cases, death. The more severe adverse events have been associated with the intravenous administration of colchicine.
5.Dosage: In prophylaxis (the most common use), the dosage of colchicine is 0.6 mg one to three times daily. For terminating a gouty attack, a regimen of 1.2 mg followed by a single 0.6-mg oral dose was as effective as higher dose regimens, and adverse events were less frequent. In 2008, the FDA requested that intravenous preparations containing colchicine be discontinued in the United States because of their potential lifethreatening adverse effects. Therefore, intravenous colchicine is no longer available.
In 2009, the FDA approved a new oral formulation of colchicine for the treatment of gout, allowing Colcrys (a branded colchicine) marketing exclusivity in the United States. Generic colchicine rather than Colcrys is available throughout the rest of the world.
NSAIDS IN GOUT
In addition to inhibiting prostaglandin synthase, NSAIDs inhibit urate crystal phagocytosis. Aspirin is not used because it causes renal retention of uric acid at low doses (≤2.6 g/d). It is uricosuric at doses greater than 3.6 g/d. Indomethacin is commonly used in the initial treatment of gout as a replacement for colchicine. For acute gout, 50 mg is given three times daily; when a response occurs, the dosage is reduced to 25 mg three times daily for 5–7 days.
All other NSAIDs except aspirin, salicylates, and tolmetin have been successfully used to treat acute gouty episodes. Oxaprozin, which lowers serum uric acid, is theoretically a good choice. These agents appear to be as effective and safe as the older drugs.
transport sites of the renal tubule (see Chapter 15). Probenecid is completely reabsorbed by the renal tubules and is metabolized slowly with a terminal serum half-life of 5–8 hours. Sulfinpyrazone or its active hydroxylated derivative is excreted by the kidneys. Even so, the duration of its effect after oral administration is almost as long as that of probenecid, which is given once or twice daily.
2.Pharmacodynamics: Uricosuric drugs—probenecid, sulfinpyrazone, as well as fenofibrate, and losartan—inhibit active transport sites for reabsorption and secretion in the proximal renal tubule so that net reabsorption of uric acid in the proximal tubule is decreased. Because aspirin in doses of less than 2.6 g daily causes net retention of uric acid by inhibiting the secretory transporter, it should not be used for analgesia in patients with gout. The secretion of other weak acids (eg, penicillin) is also reduced by uricosuric agents.
3.As the urinary excretion of uric acid increases, the size of the urate pool decreases, although the plasma concentration may not be greatly reduced. In patients who respond favorably, tophaceous deposits of urate are reabsorbed, with relief of arthritis and remineralization of bone. With the ensuing increase in uric acid excretion, a predisposition to the formation of renal stones is augmented rather than decreased; therefore, the urine volume should be maintained at a high level, and at least early in treatment, the urine pH should be kept above 6.0 by the administration of alkali.
4.Indications: Uricosuric therapy should be initiated in gouty patients with underexcretion of uric acid when allopurinol or febuxostat is contraindicated or when tophi are present. Therapy should not be started until 2–3 weeks after an acute attack.
5.Adverse Effects: Both of these organic acids cause equivalent GI irritation, but sulfinpyrazone is more active in this regard. A rash may appear after the use of either compound. Nephrotic syndrome has occurred after the use of probenecid. Both sulfinpyrazone and probenecid may rarely cause aplastic anemia.
6.Contraindications and Cautions: It is essential to maintain a large urine volume to minimize the possibility of stone formation.
7.Dosage: Probenecid is usually started at a dosage of 0.5 g orally daily in divided doses, progressing to 1 g daily after 1 week. Sulfinpyrazone is started at a dosage of 200 mg orally daily, progressing to 400–800 mg daily. It should be given in divided doses with food to reduce adverse GI effects.
URICOSURIC AGENTS
Probenecid and sulfinpyrazone are uricosuric drugs employed to decrease the body pool of urate in patients with tophaceous gout or in those with increasingly frequent gouty attacks. In a patient who excretes large amounts of uric acid, the uricosuric agents should not be used. Lesinurad (RDEA594) is a promising new uricosuric agent that is currently in phase 3 trials.
1.Chemistry and Pharmacokinetics: Uricosuric drugs are organic acids (Figure 36–6) and, as such, act at the anion
ALLOPURINOL
The preferred and standard-of-care therapy for gout during the period between acute episodes is allopurinol, which reduces total uric acid body burden by inhibiting xanthine oxidase.
1.Chemistry and Pharmacokinetics: The structure of allopurinol, an isomer of hypoxanthine, is shown in Figure 36–7. Allopurinol is approximately 80% absorbed after oral administration and has a terminal serum half-life of 1–2 hours.

662 |
SECTION VI Drugs Used to Treat Diseases of the Blood, Inflammation, & Gout |
|
|
|
|
|
|
|
|||||||||||||||||||
|
|
|
O |
|
|
|
|
|
|
|
|
O |
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
Xanthine |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
HN |
|
|
|
|
|
|
|
|
oxidase |
|
HN |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
N |
N |
|
|
O |
|
|
|
|
N |
N |
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
N |
|
|
|
|
|
|
N |
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
H |
|
|
|
|
|
H |
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
H |
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
Allopurinol |
|
|
|
Alloxanthine |
|
|
|
|
|
|
|
|
|
|
|||||||||||
|
|
|
O |
|
|
|
|
|
|
|
O |
|
|
|
|
|
O |
|
|
|
|||||||
|
|
|
|
|
|
|
|
N |
|
Xanthine |
|
|
|
|
|
|
N |
|
Xanthine |
|
|
|
|
|
N |
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
HN |
|
6 |
|
|
|
oxidase |
|
HN |
|
|
|
|
|
oxidase |
|
HN |
|
|
|
|||||||
|
1 |
5 |
|
7 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
2 |
|
|
|
4 |
|
8 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
OH |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
3 |
|
9 |
|
|
|
O |
|
|
|
|
|
|
|
O |
|
|
|
|
|
|
|
||||
|
|
|
|
|
N |
|
|
|
|
|
|
|
N |
|
|
|
|
|
|
N |
|||||||
|
|
|
N |
|
|
|
|
|
N |
|
|
|
N |
||||||||||||||
|
|
|
|
|
H |
|
|
|
|
H |
|
|
|
|
H |
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
H |
|
|
|
|
H |
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
Hypoxanthine |
|
|
|
|
Xanthine |
|
|
|
|
|
Uric acid |
|||||||||||||||
FIGURE 36 7 Inhibition of uric acid synthesis by allopurinol occurs because allopurinol and alloxanthine inhibit xanthine oxidase.
(Reproduced, with permission, from Meyers FH, Jawetz E, Goldfien A: Review of Medical Pharmacology, 7th ed. McGraw-Hill, 1980. Copyright © The McGraw-Hill Companies, Inc.)
Like uric acid, allopurinol is metabolized by xanthine oxidase, but the resulting compound, alloxanthine, retains the capacity to inhibit xanthine oxidase and has a long enough duration of action so that allopurinol is given only once a day.
2.Pharmacodynamics: Dietary purines are not an important source of uric acid. Quantitatively important amounts of purine are formed from amino acids, formate, and carbon dioxide in the body. Those purine ribonucleotides not incorporated into nucleic acids and derived from nucleic acid degradation are converted to xanthine or hypoxanthine and oxidized to uric acid (Figure 36–7). Allopurinol inhibits this last step, resulting in a fall in the plasma urate level and a decrease in the overall urate burden. The more soluble xanthine and hypoxanthine are increased.
3.Indications: Allopurinol is often the first-line agent for the treatment of chronic gout in the period between attacks and it tends to prolong the intercritical period. As with uricosuric agents, the therapy is begun with the expectation that it will be continued for years if not for life. When initiating allopurinol, colchicine or NSAID should be used until steady-state serum uric acid is normalized or decreased to less than 6 mg/dL and they should be continued for 6 months or longer. Thereafter, colchicine or the NSAID can be cautiously stopped while continuing allopurinol therapy.
4.Adverse Effects: In addition to precipitating gout (the reason to use concomitant colchicine or NSAID), GI intolerance (including nausea, vomiting, and diarrhea), peripheral neuritis and necrotizing vasculitis, bone marrow suppression, and aplastic anemia may rarely occur. Hepatic toxicity and interstitial nephritis have been reported. An allergic skin reaction characterized by pruritic maculopapular lesions occurs in 3% of patients. Isolated cases of exfoliative dermatitis have been reported. In very rare cases, allopurinol has become bound to the lens, resulting in cataracts.
5.Interactions and Cautions: When chemotherapeutic purines (eg, azathioprine) are given concomitantly with allopurinol, their dosage must be reduced by about 75%. Allopurinol may also increase the effect of cyclophosphamide. Allopurinol inhibits the metabolism of probenecid and oral anticoagulants and may increase hepatic iron concentration. Safety in children and during pregnancy has not been established.
6.Dosage: The initial dosage of allopurinol is 50–100 mg/d. It should be titrated upward until serum uric acid is below 6 mg/ dL; this level is commonly achieved at 300–400 mg/d but is not restricted to this dose; doses as high as 800 mg/d may be needed.
As noted above, colchicine or an NSAID should be given during the first months of allopurinol therapy to prevent the gouty arthritis episodes that sometimes occur.
FEBUXOSTAT
Febuxostat is a non-purine xanthine oxidase inhibitor that was approved by the FDA in 2009.
1.Pharmacokinetics: Febuxostat is more than 80% absorbed following oral administration. With maximum concentration achieved in approximately 1 hour and a half-life of 4–18 hours, once-daily dosing is effective. Febuxostat is extensively metabolized in the liver. All of the drug and its inactive metabolites appear in the urine, although less than 5% appears as unchanged drug.
2.Pharmacodynamics: Febuxostat is a potent and selective inhibitor of xanthine oxidase, thereby reducing the formation of xanthine and uric acid without affecting other enzymes in the purine or pyrimidine metabolic pathway. In clinical trials, Febuxostat at daily dosing of 80 mg or 120 mg was more