

6 семестр / Физическая химия 1-ая аттестация 6 семестр
.docx
работу
можно направить на нагрев более горячего
тела. Нарушается формулировка Клаузиуса,
так как в окружающих телах нет никаких
изменений. Замечание.
Второе
начало т/д запрещает создание вечного
двигателя второго рода, полностью
превращающего в работу всю полученную
энергию. (Вечный двигатель первого рода
совершает работу без получения
энергии).
Возрастание
энтропии при необратимых процессах:
Важной
особенностью энтропии является ее
поведение при необратимых процессах.
Для необратимого кругового процесса
справедливо соотношение:
![]() О
О но
является обобщением частного уравнения:
но
является обобщением частного уравнения:
![]() которое,
в свою очередь, является следствием
первой теоремы Карно.
Рассмотрим
процесс, при котором система необратимым
образом переходит из равновесного
состояния 1 в равновесное состояние 2
(на рис. 95 он показан сплошной линией).
Необратимость перехода означает,
что промежуточные состояния неравновесны.
Как при таком переходе изменяется
энтропия системы? Чтобы это выяснить,
вернем систему в первоначальное состояние
каким-нибудь обратимым путем, например
путем, показанным на рис. 95 пунктирной
линией. Получившийся круговой процесс
необратим, потому что одна его часть
необратима. Поэтому для него справедливо
уравнение
которое,
в свою очередь, является следствием
первой теоремы Карно.
Рассмотрим
процесс, при котором система необратимым
образом переходит из равновесного
состояния 1 в равновесное состояние 2
(на рис. 95 он показан сплошной линией).
Необратимость перехода означает,
что промежуточные состояния неравновесны.
Как при таком переходе изменяется
энтропия системы? Чтобы это выяснить,
вернем систему в первоначальное состояние
каким-нибудь обратимым путем, например
путем, показанным на рис. 95 пунктирной
линией. Получившийся круговой процесс
необратим, потому что одна его часть
необратима. Поэтому для него справедливо
уравнение![]() Но
Но Второй из двух интегралов, поскольку
он относится к обратимому процессу,
равен
Второй из двух интегралов, поскольку
он относится к обратимому процессу,
равен
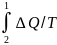 =S1-S2.
Следовательно
=S1-S2.
Следовательно
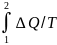 +S1-S2<0
или S2-S1>
Если система замкнута, т. е. изолирована
от источников теплоты, то dQ=0
и S2-S1>0,
или S2>S1.
Отсюда следует, что энтропия замкнутой
(т. е. адиабатно изолированной) системы
при необратимом процессе возрастает.
Можно сказать что энтропия замкнутой
системы либо остается постоянной, либо
возрастает. Полученный
закон возрастания энтропии при необратимых
процессах-одна
из важнейших особенностей величины
энтропии. Он тем более важен, что, понятие
об обратимом процессе явл. идеализацией.
Ведь при обратимом процессе система на
любой его стадии должна находиться в
состоянии т/д равновесия. Для установления
равновесия требуется время, и поэтому
процесс, чтобы быть вполне обратимым,
должен протекать бесконечно медленно,
что никогда не бывает. Для необратимых
же процессов в замкнутых системах
энтропия всегда возрастает, и это св-во
так же присуще энтропии, как энергии
свойственно сохраняться при любых
процессах в замкнутых системах. Именно
потому, что энергия обладает св-вом
сохраняться в замкнутой системе, она
(энергия) не может служить фу-цией,
показывающей, в каком направлении идут
процессы в такой системе; ведь при любом
изменении состояния энергия в начале
и в конце процесса одна и та же и она
поэтому не дает возможности отличить
друг от друга начальное и конечное
состояния. Энтропия же, в естественно
идущих процессах всегда возрастающая,
позволяет судить, какое направление
процесса возможно и какое нет, какое
состояние является начальным и какое
конечным.
Абсолютная
температура и термодинамическая шкала
t:
Абсолютная t-
это темпе-ратура по шкале Кельвина. За
0 градусов в этой шкале принимается t,
при которой все молекулы прекращают
свое тепловое движение. При этом изменение
t
на 1К соответствует изменению t
на 1 градус Сº. Температура Т была введена
вначале эмпирическим путем с помощью
газового термометра исходя из зависимости
между давлением и температурой идеального
газа. Но ур. для идеального газа справедливо
в ограниченном интервале значений
давлений и температур. Из
выражения КПД машины, работающей по
циклу Карно, следует что:
Qx/Qн=Tx/Tн.
Говоря что соотношение позволяет опытным
путем ввести новую абсолютную шкалу
температур, которая не зависит от свойств
рабочего тела и такую, что КПД для цикла
Карно будет зависеть только от новых
температур и будет выполняться равенство:
Qx/Qн=Ф(Тх,Тн)=Тх/Тн.
Рассмотрим цикл карно 1-2-5-6 с температурами
нагревателя Т1 и холодильникаТ3, состоящий
из двух «подциклов» 1-2-3-4 и 3-5-6-4 с
промежуточной температурой Т2.
+S1-S2<0
или S2-S1>
Если система замкнута, т. е. изолирована
от источников теплоты, то dQ=0
и S2-S1>0,
или S2>S1.
Отсюда следует, что энтропия замкнутой
(т. е. адиабатно изолированной) системы
при необратимом процессе возрастает.
Можно сказать что энтропия замкнутой
системы либо остается постоянной, либо
возрастает. Полученный
закон возрастания энтропии при необратимых
процессах-одна
из важнейших особенностей величины
энтропии. Он тем более важен, что, понятие
об обратимом процессе явл. идеализацией.
Ведь при обратимом процессе система на
любой его стадии должна находиться в
состоянии т/д равновесия. Для установления
равновесия требуется время, и поэтому
процесс, чтобы быть вполне обратимым,
должен протекать бесконечно медленно,
что никогда не бывает. Для необратимых
же процессов в замкнутых системах
энтропия всегда возрастает, и это св-во
так же присуще энтропии, как энергии
свойственно сохраняться при любых
процессах в замкнутых системах. Именно
потому, что энергия обладает св-вом
сохраняться в замкнутой системе, она
(энергия) не может служить фу-цией,
показывающей, в каком направлении идут
процессы в такой системе; ведь при любом
изменении состояния энергия в начале
и в конце процесса одна и та же и она
поэтому не дает возможности отличить
друг от друга начальное и конечное
состояния. Энтропия же, в естественно
идущих процессах всегда возрастающая,
позволяет судить, какое направление
процесса возможно и какое нет, какое
состояние является начальным и какое
конечным.
Абсолютная
температура и термодинамическая шкала
t:
Абсолютная t-
это темпе-ратура по шкале Кельвина. За
0 градусов в этой шкале принимается t,
при которой все молекулы прекращают
свое тепловое движение. При этом изменение
t
на 1К соответствует изменению t
на 1 градус Сº. Температура Т была введена
вначале эмпирическим путем с помощью
газового термометра исходя из зависимости
между давлением и температурой идеального
газа. Но ур. для идеального газа справедливо
в ограниченном интервале значений
давлений и температур. Из
выражения КПД машины, работающей по
циклу Карно, следует что:
Qx/Qн=Tx/Tн.
Говоря что соотношение позволяет опытным
путем ввести новую абсолютную шкалу
температур, которая не зависит от свойств
рабочего тела и такую, что КПД для цикла
Карно будет зависеть только от новых
температур и будет выполняться равенство:
Qx/Qн=Ф(Тх,Тн)=Тх/Тн.
Рассмотрим цикл карно 1-2-5-6 с температурами
нагревателя Т1 и холодильникаТ3, состоящий
из двух «подциклов» 1-2-3-4 и 3-5-6-4 с
промежуточной температурой Т2.
 Энтропия
как функция состояния Энтропия,
функция состояния (S) т/д системы,
изменение которой (dS) для бесконечно
малого обратимого изменения состояния
системы равно отношению кол-ва теплоты
(δQ), полученного
системой в этом процессе (или отнятого
от системы), к абсолютной температуре (Т):
dS
= δQ/T.
Энтропия
как функция состояния Энтропия,
функция состояния (S) т/д системы,
изменение которой (dS) для бесконечно
малого обратимого изменения состояния
системы равно отношению кол-ва теплоты
(δQ), полученного
системой в этом процессе (или отнятого
от системы), к абсолютной температуре (Т):
dS
= δQ/T.
Величина dS явл.
полным дифференциалом, т.е. ее интегрирование
по любому произвольно выбранному пути
дает разность между значениями энтропии в
начальном (А) и конечном (В) состояниях:
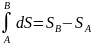 Теплота
не явл. фу-цией состояния, поэтому
интеграл от δQ зависит от выбранного
пути перехода между состояниями А и
В. Энтропия измеряется в
Дж/(моль·град).
Теплота
не явл. фу-цией состояния, поэтому
интеграл от δQ зависит от выбранного
пути перехода между состояниями А и
В. Энтропия измеряется в
Дж/(моль·град).
Понятие энтропии как функции состояния системы постулируется вторым началом термодинамики, которое выражает через энтропию различие между необратимыми и обратимыми процессами. Для первых dS>δQ/T для вторых dS=δQ/T.
Энтропия как
функция внутренней
энергии U системы,
объема V и числа молей ni i-го
компонента представляет собой
характеристическую функцию. Это явл.
следствием первого и второго начал
термодинамики и записывается уравнением:
dS=(1/T)dU+(p/T)dV-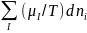 ,
где р - давление,
μi - химический потенциал i-го
компонента. Производные энтропии по
естественным переменным U, V и ni равны:
(
,
где р - давление,
μi - химический потенциал i-го
компонента. Производные энтропии по
естественным переменным U, V и ni равны:
(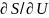 )V,
=
1/T;
(
)V,
=
1/T;
( )U,
=
p/T;
(
)U,
=
p/T;
( )V,U,
)V,U,
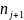 =
-
/T;
=
-
/T;
Простые
формулы связывают энтропию с
теплоемкостями при постоянном
давлении Ср и постоянном объеме Cv:
Ср=Т( Т)Р;
СV=Т(
Т)V;
Т)Р;
СV=Т(
Т)V;
С помощью энтропии формулируются условия достижения т/д равновесия системы при постоянстве ее внутренней энергии, объема и числа молей i-го компонента (изолированная система) и условие устойчивости такого равновесия:
(dS)U, V, = 0; (d2S)U, V, ,< 0; Это означает, что энтропия изолированной системы достигает максимума в состоянии т/д равновесия. Самопроизвольные процессы в системе могут протекать только в направлении возрастания энтропии.
Р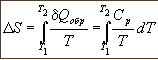 асчеты
изменения энтропии при различных
процессах
dS=
Qобр/Т,
т/д расчеты основаны на этой формуле и
на свойствах частных производных
энтропии по т/д параметрам: (
S/
T)p=Cp/T
ур-е 1, (
S/
T)v=Cv/T,
(
S/
p)т=-(
V/
T)p,
(
S/
V)т=-(
p/
T)v
Последние два тождества представляют
собой соотношения Максвелла.1)
Нагревание или охлаждение при р=const:
количество теплоты, необходимое для
изменения температуры системы, выражают
с помощью теплоемкости: δQобр = Cp dT.
(2)
Если
теплоемкость не зависит от температуры
в интервале от T1 до T2, то уравнение 1
можно проинтегрировать:∆S=Cpln(T2/T1)
(3). Если изменение температуры происходит
при постоянном объеме, то в формулах
(2) и (3) Cp надо заменить на Cv. 2)
Изотермическое расширение или сжатие.
Для расчета энтропии в этом случае надо
знать уравнение с
асчеты
изменения энтропии при различных
процессах
dS=
Qобр/Т,
т/д расчеты основаны на этой формуле и
на свойствах частных производных
энтропии по т/д параметрам: (
S/
T)p=Cp/T
ур-е 1, (
S/
T)v=Cv/T,
(
S/
p)т=-(
V/
T)p,
(
S/
V)т=-(
p/
T)v
Последние два тождества представляют
собой соотношения Максвелла.1)
Нагревание или охлаждение при р=const:
количество теплоты, необходимое для
изменения температуры системы, выражают
с помощью теплоемкости: δQобр = Cp dT.
(2)
Если
теплоемкость не зависит от температуры
в интервале от T1 до T2, то уравнение 1
можно проинтегрировать:∆S=Cpln(T2/T1)
(3). Если изменение температуры происходит
при постоянном объеме, то в формулах
(2) и (3) Cp надо заменить на Cv. 2)
Изотермическое расширение или сжатие.
Для расчета энтропии в этом случае надо
знать уравнение с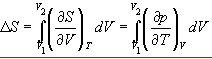 остояния
системы. Расчет основан на использовании
соотношения Максвелла:
В
частности, для изотермического расширения
идеального газа (p=nRT/V): ∆S=nRln(V2/V1).
Этот
же можно получить, если использовать
выражение для теплоты изотермич.
обратимого расширения идеального газа:
Qобр=nRT ln(V2/V1).
3) Фазовые переходы. При
обратимом фазовом переходе температура
остается постоянной, а теплота фазового
перехода при постоянном давлении равна
∆Hфп, поэтому изменение энтропии равно:
остояния
системы. Расчет основан на использовании
соотношения Максвелла:
В
частности, для изотермического расширения
идеального газа (p=nRT/V): ∆S=nRln(V2/V1).
Этот
же можно получить, если использовать
выражение для теплоты изотермич.
обратимого расширения идеального газа:
Qобр=nRT ln(V2/V1).
3) Фазовые переходы. При
обратимом фазовом переходе температура
остается постоянной, а теплота фазового
перехода при постоянном давлении равна
∆Hфп, поэтому изменение энтропии равно:
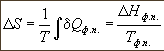 При
плавлении и кипении теплота поглощается,
поэтому энтропия в этих процессах
возрастает: Sтв < Sж < Sг. При этом
энтропия окружающей среды уменьшается
на величину ∆Sф.п., поэтому изменение
энтропии Вселенной равно 0, как и
полагается для обратимого процесса в
изолированной системе.
4) Смешение
идеальных газов при постоянных температуре
и давлении. Если n1 молей одного газа,
занимающего объем V1, смешиваются с n2
молями другого газа, занимающего объем
V2, то общий объем будет равен V1+V2, причем
газы расширяются независимо друг от
друга и общее изменение энтропии равно
сумме изменений энтропии каждого газа:
При
плавлении и кипении теплота поглощается,
поэтому энтропия в этих процессах
возрастает: Sтв < Sж < Sг. При этом
энтропия окружающей среды уменьшается
на величину ∆Sф.п., поэтому изменение
энтропии Вселенной равно 0, как и
полагается для обратимого процесса в
изолированной системе.
4) Смешение
идеальных газов при постоянных температуре
и давлении. Если n1 молей одного газа,
занимающего объем V1, смешиваются с n2
молями другого газа, занимающего объем
V2, то общий объем будет равен V1+V2, причем
газы расширяются независимо друг от
друга и общее изменение энтропии равно
сумме изменений энтропии каждого газа:
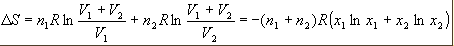 где
xi-мольная доля i-го газа в полученной
газовой смеси. Изменение энтропии всегда
положительно, т.к. все ln xi < 0, поэтому
идеальные газы всегда смешиваются
необратимо. Если при тех же условиях
смешиваются 2 порции одного и того же
газа, то ур-ие уже неприменимо. Никаких
изменений в системе при смешивании не
происходит, и ∆S=0. Тем не менее, формула
не содержит никаких индивидуальных
парам. газов, поэтому должна быть
применима и к смешению одинаковых газов.
Это противоречие-парадокс Гиббса.
Правило
Трутона. Молярная
энтропия испарения разных ве-вв при
нормальной t
кипения явл. постоянной величиной и
равняется константе Трутона:∆S
=∆H
/T
=Ktrouton,
Ktrouton
=87-88Дж/моль*К.
Гипотеза
Нернста получила название тепловая
теорема Нернста: вблизи абсолютного
нуля для конденсированных систем:
где
xi-мольная доля i-го газа в полученной
газовой смеси. Изменение энтропии всегда
положительно, т.к. все ln xi < 0, поэтому
идеальные газы всегда смешиваются
необратимо. Если при тех же условиях
смешиваются 2 порции одного и того же
газа, то ур-ие уже неприменимо. Никаких
изменений в системе при смешивании не
происходит, и ∆S=0. Тем не менее, формула
не содержит никаких индивидуальных
парам. газов, поэтому должна быть
применима и к смешению одинаковых газов.
Это противоречие-парадокс Гиббса.
Правило
Трутона. Молярная
энтропия испарения разных ве-вв при
нормальной t
кипения явл. постоянной величиной и
равняется константе Трутона:∆S
=∆H
/T
=Ktrouton,
Ktrouton
=87-88Дж/моль*К.
Гипотеза
Нернста получила название тепловая
теорема Нернста: вблизи абсолютного
нуля для конденсированных систем:
![]() Ур-ие
явл. выражением тепловой теоремы Нернста.
Знание этой теоремы и температурной
зависимости теплот р-ии позволяет
теоретически вычислить мax.
работу р-ии. Нернст
сформулировал теорему для изолированных
систем, а затем Планк распространил ее
на случай любых систем, находящихся в
термодинамич. равновесии. Как первое и
второе начала термодинамики, теорема
Нернста может рассматр. как результат
обобщения опытных фактов, поэтому ее
часто называют третьим
началом термодинамики. Иногда его
формулируют следующим образом:
энтропия любой равновесной системы при
абсолютном 0 температуры может быть
равна нулю. Отсюда следует, что при T→0
интеграл
Ур-ие
явл. выражением тепловой теоремы Нернста.
Знание этой теоремы и температурной
зависимости теплот р-ии позволяет
теоретически вычислить мax.
работу р-ии. Нернст
сформулировал теорему для изолированных
систем, а затем Планк распространил ее
на случай любых систем, находящихся в
термодинамич. равновесии. Как первое и
второе начала термодинамики, теорема
Нернста может рассматр. как результат
обобщения опытных фактов, поэтому ее
часто называют третьим
началом термодинамики. Иногда его
формулируют следующим образом:
энтропия любой равновесной системы при
абсолютном 0 температуры может быть
равна нулю. Отсюда следует, что при T→0
интеграл
 сходится на нижнем пределе, т.е. имеет
конечное значение S(0)=const или S(0)=0, причем
равенство нулю рассматривается как
наиболее вероятное. А нулевое значение
энтропии (меры беспорядка) соответствует
отсутствию теплового движения при
абсолютном нуле. При T=0 внутренняя
энергия и тепловая функция системы
прекращают зависеть от температуры,
кроме того, используя метод термодинамических
функций, можно показать, что, при T=0, от
температуры не зависит коэффициент
объемного расширения, термический
коэффициент давления и другие параметры
системы. Согласно классическим
представлениям при абсолютном нуле
возможно непрерывное множество
микросостояний системы.
Температурн.
зависимость энтропии и вычисление
энтропии из эксперимент. данных:
Расчет
изменения энтропии в разлиных процессах
основан на использовании соотношения
dS
δQ/T
и частных производных энртопии по т/д
переменным:
сходится на нижнем пределе, т.е. имеет
конечное значение S(0)=const или S(0)=0, причем
равенство нулю рассматривается как
наиболее вероятное. А нулевое значение
энтропии (меры беспорядка) соответствует
отсутствию теплового движения при
абсолютном нуле. При T=0 внутренняя
энергия и тепловая функция системы
прекращают зависеть от температуры,
кроме того, используя метод термодинамических
функций, можно показать, что, при T=0, от
температуры не зависит коэффициент
объемного расширения, термический
коэффициент давления и другие параметры
системы. Согласно классическим
представлениям при абсолютном нуле
возможно непрерывное множество
микросостояний системы.
Температурн.
зависимость энтропии и вычисление
энтропии из эксперимент. данных:
Расчет
изменения энтропии в разлиных процессах
основан на использовании соотношения
dS
δQ/T
и частных производных энртопии по т/д
переменным:
 -
4.21а
-4.21б
-4.21в.
Последние 2 тождества-соотношения
Максвела.
1.Нагревание
или охлаждение при постоянном давлении:
количество теплоты, необход. для изменения
t
системы выражают с помощью теплоемкости:
δQобр.=СрdT
-4.22.
Если теплоемкость не зависит от
температуры в интервале от Т1 до Т2 то
ур-е 4.22 можно проинтегрировать:
S=Cp*ln*(T2/T1)-4.23а.
При более сложной температурной
зависимости теплоемкости в выражении
4.23а появляются доп. слагаемые. Например,
если теплоемкость описывается рядом
Ср=а+bT+c/T
то соответствующее изменение энтропии:
-
4.21а
-4.21б
-4.21в.
Последние 2 тождества-соотношения
Максвела.
1.Нагревание
или охлаждение при постоянном давлении:
количество теплоты, необход. для изменения
t
системы выражают с помощью теплоемкости:
δQобр.=СрdT
-4.22.
Если теплоемкость не зависит от
температуры в интервале от Т1 до Т2 то
ур-е 4.22 можно проинтегрировать:
S=Cp*ln*(T2/T1)-4.23а.
При более сложной температурной
зависимости теплоемкости в выражении
4.23а появляются доп. слагаемые. Например,
если теплоемкость описывается рядом
Ср=а+bT+c/T
то соответствующее изменение энтропии:
![]() 4.23б.
Если изменения t
происходят при V=const,
то в формулах 4.22 и 4.23 Ср заменяем на
Сv.
2.
Изотермическое расширение или сжатие:Нужно
знать ур-е состояния системы, расчет
основан на соотношении Максвела:
-4.24.
В частности при изотермическом расширении
идеального газа (р=nRT/V):
∆S=nRln(V2/V1)-4.25
Этот же результат можно получить если
использовать выражение для теплоты
изотермического обратимого расширения
идеального газа: Qобр.=nRTln(V2/V1).
3.
Фазовые переходы: при
обратимом фазовом переходе температура
остается постоянной, теплота фазового
перехода при постоянном давлении равна
∆фпН поэтому изменение энтропии
составляет:
4.23б.
Если изменения t
происходят при V=const,
то в формулах 4.22 и 4.23 Ср заменяем на
Сv.
2.
Изотермическое расширение или сжатие:Нужно
знать ур-е состояния системы, расчет
основан на соотношении Максвела:
-4.24.
В частности при изотермическом расширении
идеального газа (р=nRT/V):
∆S=nRln(V2/V1)-4.25
Этот же результат можно получить если
использовать выражение для теплоты
изотермического обратимого расширения
идеального газа: Qобр.=nRTln(V2/V1).
3.
Фазовые переходы: при
обратимом фазовом переходе температура
остается постоянной, теплота фазового
перехода при постоянном давлении равна
∆фпН поэтому изменение энтропии
составляет:
 -4.26.
При плавлении и кипении теплота
поглощается, поэтому энтропия в этих
процессах возрастает: Sтв < Sж < Sг. При
этом энтропия окружающей среды уменьшается
на величину ∆ф.п.S, поэтому изменение
энтропии Вселенной равно 0, как и
полагается для обратимого процесса в
изолированной системе. Если фазовый
переход происходит при температуре,
отличной от температуры обратимого
фазового перехода, то использовать
формулу (4.26) нельзя, так как согласно
второму началу термодинамики при
необратимых процессах dS>δQ/Tфп
-
4.27. В таких случаях для расчета энтропии
используют ее свойства как функции
состояния, рассматривая цикл:
-4.26.
При плавлении и кипении теплота
поглощается, поэтому энтропия в этих
процессах возрастает: Sтв < Sж < Sг. При
этом энтропия окружающей среды уменьшается
на величину ∆ф.п.S, поэтому изменение
энтропии Вселенной равно 0, как и
полагается для обратимого процесса в
изолированной системе. Если фазовый
переход происходит при температуре,
отличной от температуры обратимого
фазового перехода, то использовать
формулу (4.26) нельзя, так как согласно
второму началу термодинамики при
необратимых процессах dS>δQ/Tфп
-
4.27. В таких случаях для расчета энтропии
используют ее свойства как функции
состояния, рассматривая цикл:
 4.
Смешение идеальных газов при постоянных
t
и давлении:Если
n1 молей одного газа, занимающего объем
V1, смешиваются с n2 молями другого газа,
занимающего объем V2, то общий объем
будет равен V1 + V2, причем газы расширяются
независимо друг от друга и занимают
весь объем, поэтому общее изменение
энтропии равно сумме изменений энтропии
каждого газа:
-4.28.
где xi – мольная доля i-го газа в полученной
газовой смеси. Изменение энтропии (4.28)
всегда положительно, т.к. все ln xi<0,поэтому
идеальные газы всегда смешиваются
необратимо.Если при тех же условиях
смешиваются две порции одного и того
же газа, то уравнение (4.28) уже неприменимо.
Никаких изменений в системе при смешивании
не наблюдается, и ∆S=0. Тем не менее,
формула (4.28) не содержит никаких
индивидуальных параметров газов,
поэтому, казалось бы, должна быть
применима и к смешению одинаковых газов.
Это противоречие называют парадоксом
Гиббса.
5.
Изменение энтропии при химич. р-ях:
Разность
мольных энтропий продуктов р-ии и
реагентов, взятых в стандартных
состояниях, называется стандартной
энтропией реакции. Для реакции
4.
Смешение идеальных газов при постоянных
t
и давлении:Если
n1 молей одного газа, занимающего объем
V1, смешиваются с n2 молями другого газа,
занимающего объем V2, то общий объем
будет равен V1 + V2, причем газы расширяются
независимо друг от друга и занимают
весь объем, поэтому общее изменение
энтропии равно сумме изменений энтропии
каждого газа:
-4.28.
где xi – мольная доля i-го газа в полученной
газовой смеси. Изменение энтропии (4.28)
всегда положительно, т.к. все ln xi<0,поэтому
идеальные газы всегда смешиваются
необратимо.Если при тех же условиях
смешиваются две порции одного и того
же газа, то уравнение (4.28) уже неприменимо.
Никаких изменений в системе при смешивании
не наблюдается, и ∆S=0. Тем не менее,
формула (4.28) не содержит никаких
индивидуальных параметров газов,
поэтому, казалось бы, должна быть
применима и к смешению одинаковых газов.
Это противоречие называют парадоксом
Гиббса.
5.
Изменение энтропии при химич. р-ях:
Разность
мольных энтропий продуктов р-ии и
реагентов, взятых в стандартных
состояниях, называется стандартной
энтропией реакции. Для реакции
 стандартная
энтропия реакции равна разности
абсолютных энтропий продуктов и реагентов
с учетом стехиометрических коэффициентов:
стандартная
энтропия реакции равна разности
абсолютных энтропий продуктов и реагентов
с учетом стехиометрических коэффициентов:
![]() Для
расчета абсолютной энтропии в-тв в
стандартном состоянии надо знать
зависимости теплоемкости Cp от температуры
для каждой из фаз, а также температуры
и энтальпии фп. Так, например, абсолютная
энтропия газообразного в-ва в стандартном
состоянии при температуре T складывается
из следующих составляющих:
-4.30
рис
4.2 (а) к расчету энтропии по результ.
измерения теплоемкости; (б) зависимость
энтропии от температуры. В т/д таблицах
обычно приводят значения абсолютной
энтропии в станд состоянии при температуре
298 К или табулируют значения Sº с шагом
по температуре 100 К. Абсолютные энтропии
участников реакции при давлении, отличном
от стандартного, находят, интегрируя
соотношение (4.21.б):
Для
расчета абсолютной энтропии в-тв в
стандартном состоянии надо знать
зависимости теплоемкости Cp от температуры
для каждой из фаз, а также температуры
и энтальпии фп. Так, например, абсолютная
энтропия газообразного в-ва в стандартном
состоянии при температуре T складывается
из следующих составляющих:
-4.30
рис
4.2 (а) к расчету энтропии по результ.
измерения теплоемкости; (б) зависимость
энтропии от температуры. В т/д таблицах
обычно приводят значения абсолютной
энтропии в станд состоянии при температуре
298 К или табулируют значения Sº с шагом
по температуре 100 К. Абсолютные энтропии
участников реакции при давлении, отличном
от стандартного, находят, интегрируя
соотношение (4.21.б):
 -4.31
Постулат
Планка: Абсолютное
значение энтропии, полученное при
интегрировании dS
δQ/T
для обратимых процессов, известно с
точностью до постоянной интегрирования
(S0):
-4.31
Постулат
Планка: Абсолютное
значение энтропии, полученное при
интегрировании dS
δQ/T
для обратимых процессов, известно с
точностью до постоянной интегрирования
(S0):
 +S0.
Значение
этой постоянной устанавливается третьим
законом термодинамики:*При
нулевой абсолютной температ. энтропия
любых веществ, находящихся в равновесном
состоянии, имеет одно и то же значение,
не зависящее от фаз. состояния ве-ва. В
изотермических процессах, происходящих
при T=0, энтропия не зависит ни от обобщенных
сил, ни от обобщенных координат.Так как
при 0К энтропия всех ве-в одинакова, то
конкретное значение S0 несущественно и
его можно принять равным 0 (постулат
Планка):*При
абсолютном нуле все идеальные кристаллы
имеют одинаковую энтропию, равную 0.
Постулат Планка позволяет ввести понятие
абсолютной энтропии в-ва, т.е. энтропии,
отсчитанной от нулевого значения при
T=0. Используя
понятие абсолютной энтропии, следует
помнить, что вывод о постоянстве S0
относится лишь к полностью равновесным
при 0 К системам. На самом деле при
понижении температуры ↓ скорости
релаксационных процессов, и в веществе
может «замораживаться» некоторая
остаточная энтропия.Справедливость
постулата Планка подтверждается
статистическим толкованием понятия
«энтропия». Статистическое определение
энтропии основано на идее о том, что
необратимые процессы в термодинамике
вызваны переходом системы в более
вероятное состояние, поэтому энтропию
можно связать с вероятностью:S=klnW-1.
где k–постоянная Больцмана (k=R/NA),
W–термодинамич. вероятность, т.е. число
микросостояний, которые соответствуют
данному макросостоянию системы. Формулу
(1)
называют формулой Больцмана. С ↑ кол-ва
молекул и числа доступных уровней
энергии термодинамич. вероятность резко
↑, так что для обычных молекулярных
систем при ↑ температуры и разупорядоченности
энтропия ↑. Верно и обратное.
+S0.
Значение
этой постоянной устанавливается третьим
законом термодинамики:*При
нулевой абсолютной температ. энтропия
любых веществ, находящихся в равновесном
состоянии, имеет одно и то же значение,
не зависящее от фаз. состояния ве-ва. В
изотермических процессах, происходящих
при T=0, энтропия не зависит ни от обобщенных
сил, ни от обобщенных координат.Так как
при 0К энтропия всех ве-в одинакова, то
конкретное значение S0 несущественно и
его можно принять равным 0 (постулат
Планка):*При
абсолютном нуле все идеальные кристаллы
имеют одинаковую энтропию, равную 0.
Постулат Планка позволяет ввести понятие
абсолютной энтропии в-ва, т.е. энтропии,
отсчитанной от нулевого значения при
T=0. Используя
понятие абсолютной энтропии, следует
помнить, что вывод о постоянстве S0
относится лишь к полностью равновесным
при 0 К системам. На самом деле при
понижении температуры ↓ скорости
релаксационных процессов, и в веществе
может «замораживаться» некоторая
остаточная энтропия.Справедливость
постулата Планка подтверждается
статистическим толкованием понятия
«энтропия». Статистическое определение
энтропии основано на идее о том, что
необратимые процессы в термодинамике
вызваны переходом системы в более
вероятное состояние, поэтому энтропию
можно связать с вероятностью:S=klnW-1.
где k–постоянная Больцмана (k=R/NA),
W–термодинамич. вероятность, т.е. число
микросостояний, которые соответствуют
данному макросостоянию системы. Формулу
(1)
называют формулой Больцмана. С ↑ кол-ва
молекул и числа доступных уровней
энергии термодинамич. вероятность резко
↑, так что для обычных молекулярных
систем при ↑ температуры и разупорядоченности
энтропия ↑. Верно и обратное.