

Химические свойства фенолов Кислотные свойства фенолов
Несмотря на то, что фенолы по строению подобны спиртам, они являются намного более сильными кислотами, чем спирты. Вместе с тем делокализация заряда в феноксид-ионе происходит в меньшей степени, чем в карбоксилат-ионе, соответственно фенолы более слабые кислоты по сравнению с карбоновыми кислотами. Фенолы растворяются в водном растворе гидроксида натрия, но они не реагируют c гидрокарбонатом натрия. Это простейший, хотя и не очень надежный тест, по которому можно различать фенолы и карбоновые кислоты, которые взаимодействуют c гидрокарбонатом натрия c выделением углекислого газа. Влияние заместителя в бензольном кольце на кислотность фенолов согласуется с представлениями об их электронных эффектах. Электронодонорные заместители понижают, a электроноакцепторные - усиливают кислотные свойства фенолов. Фенолы диссоциируют в водных растворах с образованием фенолят-ионов и ионов водорода:

В отличие от спиртов, фенолы реагируют не только с щелочными и щелочноземельными металлами, но и с растворами щелочей, образуя феноляты:

С увеличением длины углеводородного радикала скорость этой реакции замедляется. В присутствии следов влаги образующиеся алкоголяты разлагаются до исходных спиртов.
Таутомерия фенолов
Между амбидентными феноксид- и енолят-ионами существует определенная аналогия. Фенол также является аналогом енола и между ним и его кето-формами (2,4- и 2,5-циклогексадиенами) должны существовать отношения, подобные тем, которые наблюдаются для равновесия кето- и енольной форм кетонов.
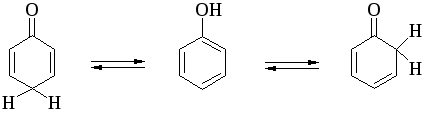
Соотношение двух таутомерных форм здесь полностью обратно тому, которое наблюдается для кетонов, где преобладает кето-форма. Устойчивость таутомерных кето-форм возрастает при переходе к полиатомным фенолам. Так, при плавлении 1,4-дигидроксинафталина получается равновесная смесь, содержащая 10%-дикетоформы.

В 1968 году В.А.Коптюг с сотрудниками предложил простой и чрезвычайно эффективный способ стабилизации кето-формы разнообразных фенолов с помощью сильных кислот Льюиса - хлорида или бромида алюминия. Эти жесткие кислоты Льюиса связывают жесткий карбонильный кислород кето-формы в очень стабильный комплекс, который может быть зафиксирован. Кето-енольная таутомерия лежит в основе замещения фенольного гидроксила на аминогруппу, которое происходит при нагревании 1- или 2-гидроксинафталина, сульфопроизводных α- и β-нафтолов, 6- или 8-гидроксихинолинов и других гидроксипроизводных нафталина, антрацена, хинолина с водным раствором сульфита или гидросульфита аммония при 130-150оС.

2.3 Этерификация фенолов
Ариловые эфиры карбоновых кислот получают ацилированием фенолов или их Na-, K-солей галогенангидридами или ангидридами кислот.


Реакции электрофильного замещения в ароматическом кольце
Гидроксильная группа относится к числу групп, активирующих электрофильное замещение в ароматическом кольце и направляющих заместитель в орто- и пара- положения. Активирующее влияние гидроксильной группы настолько сильно, что в отдельных случаях реакцию трудно остановить на стадии введения только одного заместителя. Фенолы вступают практически во все типичные реакции электрофильного замещения как с сильными, так и со слабыми электрофильными агентами.
Галогенирование фенолов
Галогенирование фенолов не требует катализа кислотами Льюиса (FeCl3, FeBr3, AlCl3 и др.) и легко осуществляется под действием молекулярного галогена. Галогенирование фенола молекулярным бромом или хлором в полярной среде практически невозможно остановить на стадии моногалогенирования, поскольку реагирующей частицей здесь является фенолят-ион. Фенолят-ион содержит очень сильную активирующую группу - анион кислорода и скорость галогенирования фенолят-иона по крайней мере в тысячу раз выше, чем фенола. Галогензамещенный фенол является более сильной кислотой, чем фенол, он легче диссоциирует, что облегчает введение второго и третьего атома галогена в орто- и пара-положения.
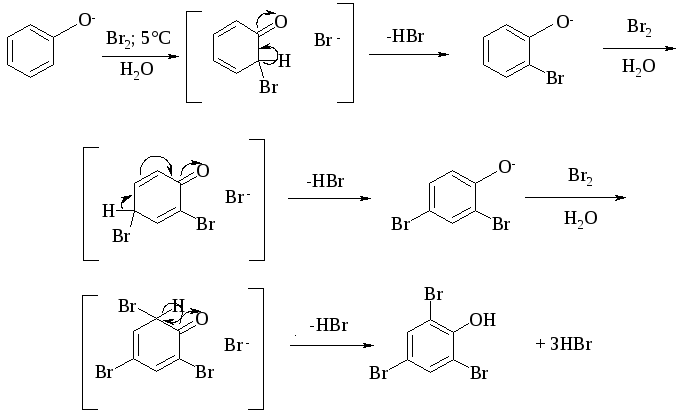
При бромировании фенола в растворе бромистоводородной кислоты или при хлорировании в соляной кислоте диссоциация полностью подавляется и галогенированию подвергается сам фенол. При этом в зависимости от условий и количества галогена может быть получен п-бромфенол или 2,4-дибромфенол.


Аналогичным образом протекает и хлорирование фенола, но здесь получается значительное количество о-хлорфенола. Моногалогензамещенные производные фенолов удобно получать при галогенировании в неполярной среде, что также исключает диссоциацию фенолов.


Во всех случаях соотношение пара- и орто-изомеров при бромировании и иодировании значительно выше, чем при хлорировании.