

Катализаторы синтеза аммиака.
Синтез аммиака протекает с заметной скоростью только в присутствии катализатора, причем катализаторами данной реакции служат твердые вещества. Гетерогенно-каталитический процесс синтеза аммиака имеет сложный механизм, который может быть описан следующими стадиями:
1) диффузия молекул азота и водорода к поверхности катализатора;
2) хемосорбция молекул реагентов на поверхности катализатора;
3) поверхностная химическая реакция с образованием неустойчивых промежуточных комплексов и взаимодействие между ними;
4) десорбция продукта;
5) диффузия продукта реакции (аммиака) от поверхности катализатора в газовую фазу.
Исследование кинетики и механизма реакции синтеза позволило сделать вывод, что лимитирующей стадией процесса является хемосорбция азота.
В промышленности нашли применение железные катализаторы, получаемые сплавлением оксидов железа с активаторами (промоторами) и последующими восстановлением оксидов железа. В качестве активаторов применяются оксиды кислотного и амфотерного характера — Al2О3. SiО2. TiО2 и др., а также оксиды щелочных и щелочноземельных металлов — К2О, Na2O, CaO, MgO и др.
Активность катализатора, его структура и состав поверхности в значительной степени определяются условиями восстановления. Процесс восстановления катализатора можно описать суммарным уравнением:
Fe3O4 + 4H2 → 3Fe + 4H2O; H>0
Катализатора синтеза аммиака необратимо отравляются сернистыми соединениями и хлором. Их концентрация в газе в сумме не должна превышать 5-10%.
Соединения (Н2О, СО, СО2) и кислород, присутствующие в азотоводородной смеси, являются сильными каталитическими ядами, снижающими активность катализатора обратимо. Их отравляющее действие пропорционально содержанию в них кислорода. Если в составе свежего газа имеются кислородосодержащие примеси или масло, газ следует вводить в цикл перед вторичной конденсацией для удаления вредных соединений конденсирующимся NH3.
Выделение аммиака:
Чтобы выделить аммиак, азотаводородную смесь вместе с аммиаком охлаждают до температуры сжижения аммиака. Достичь полной конденсации аммиака не удается; наибольшая часть его остается в азотоводородной смеси.
При давлении 30 МПа газ необходимо охлаждать до -5°С. Для этого помимо водяного или воздушного охлаждения азотоводородную смесь охлаждают кипящим жидким аммиаком. В системах, работающих при более высоком давлении (например, 60-100 МПа), для выделения аммиака из азотоводородной смеси можно ограничиться воздушным или водяным охлаждением. Непрореагировавшая азотоводородная смесь с остаточным аммиаком вновь возвращается на синтез аммиака.
О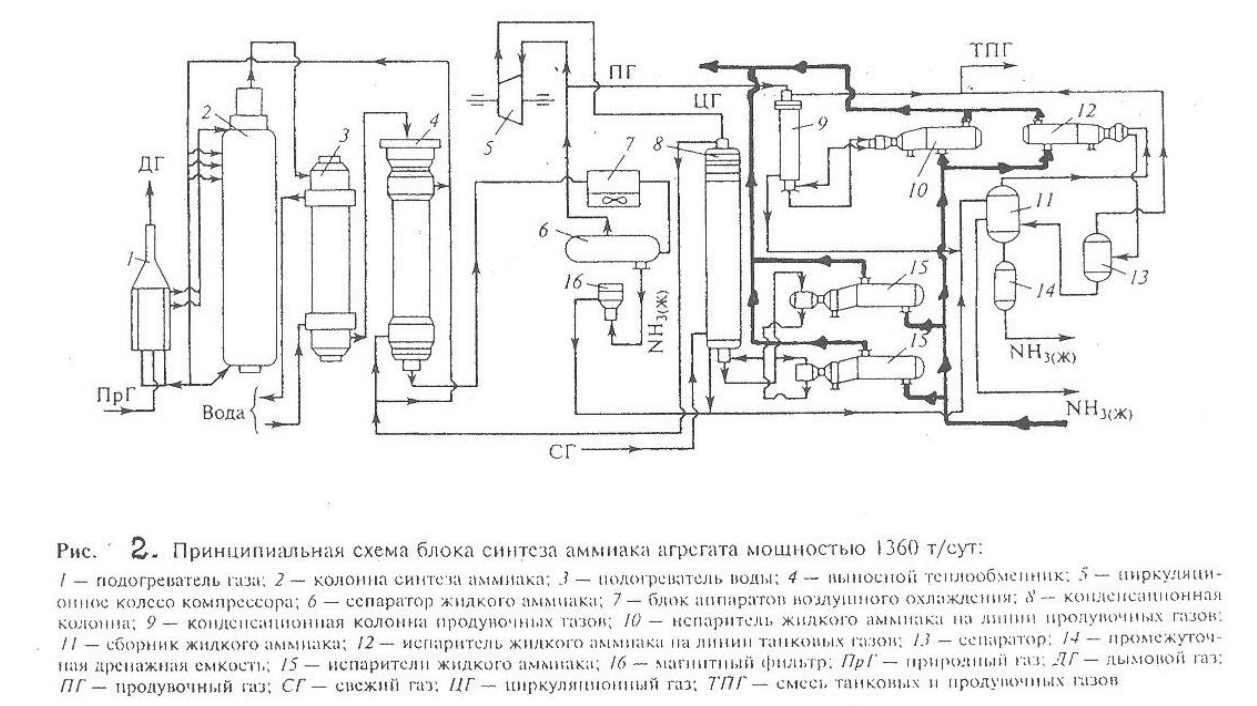 писание
технологической схемы процесса.
писание
технологической схемы процесса.

Принципиальная технологическая схема синтеза аммиака в агрегате мощность 1360 т/сут.
Свежая азотоводородная смесь (СГ) после очистки метанированием сжимается в центробежном компрессоре до давления 32 МПа и после охлаждения в воздушном холодильнике (на схеме не показан) поступает в нижнюю часть конденсационной колонны 8 для очистки от остаточных примесей СО2, Н2О и следов масла. Свежий газ барботирует через слой сконденсировавшегося жидкого аммиака, освобождается при этом от водяных паров, следов СО2 и масла, насыщается аммиаком до 3-5% и смешивается с циркуляционным газом. Полученная смесь проходит по трубкам теплообменника конденсационной колонны и направляется в межтрубное пространство выносного теплообменника 4, где нагревается до 185-195°С за счет теплоты газа, выходящего из колонны синтеза. Затем циркуляционный газ поступает в колонны синтеза 2.
В колонне синтеза газ проходит снизу
вверх
по кольцевой
щели
между корпусом колонны и кожухом насадки
и поступает в межтрубное
пространство
внутреннего
теплообменника,
размещенного в горловине корпуса колонны
синтеза. В теплообменнике циркуляционный
газ нагревается до температуры
начала реакции 400-440°С
за счет теплоты конвертированного газа
и затем последовательно проходит четыре
слоя катализатора, в результате чего
концентрация аммиака в газе повышается
до 15%.
Пройдя через центральную трубу, при
температуре 500-515°С
азото-водородно-аммиачная смесь
направляется во внутренний
теплообменник,
где охлаждается до 330°С.
Дальнейшее охлаждение газовой смеси
до 215°С
осуществляется в трубном пространстве
подогревателя питательной воды 3, в
трубном пространстве выносного
теплообменника 4 до
65°С
за счет холодного циркулирующего газа,
идущего по межтрубному пространству,
и затем в аппаратах воздушного охлаждения
7
до 40°С,
при этом часть аммиака конденсируется.
Жидкий аммиак, сконденсировавшийся при
охлаждении, отделяется в сепараторе
6,
а затем смесь, содержащая 10-12% NH3,
идет на циркуляционное колесо компрессора
5
азотоводородной смеси, где сжимается
до 32 МПа. Циркуляционный газ при
температуре 50°С
поступает в систему вторичной конденсации,
включающую конденсационную
колонну 8
и испарители
жидкого аммиака 15.
В конденсационной колонне газ охлаждается
до 18°С
и в испарителе за счет кипения аммиака
в межтрубном пространстве до —
5°С.
Из трубного пространства испарителей
смесь охлажденного циркуляционного
газа и сконденсировавшегося аммиака
поступает в сепарационную часть
конденсационной колонны, где происходит
отделение
жидкого аммиака
от газа
и смешение свежей азотоводородной смеси
с циркуляционным газом. Далее газовая
смесь проходит корзину с фарфоровыми
кольцами Рашига, где отделяется от
капель жидкого аммиака, поднимается по
трубкам теплообменника и направляется
в выносной теплообменник 4, а затем в
колонну синтеза.
колонне синтеза газ проходит снизу
вверх
по кольцевой
щели
между корпусом колонны и кожухом насадки
и поступает в межтрубное
пространство
внутреннего
теплообменника,
размещенного в горловине корпуса колонны
синтеза. В теплообменнике циркуляционный
газ нагревается до температуры
начала реакции 400-440°С
за счет теплоты конвертированного газа
и затем последовательно проходит четыре
слоя катализатора, в результате чего
концентрация аммиака в газе повышается
до 15%.
Пройдя через центральную трубу, при
температуре 500-515°С
азото-водородно-аммиачная смесь
направляется во внутренний
теплообменник,
где охлаждается до 330°С.
Дальнейшее охлаждение газовой смеси
до 215°С
осуществляется в трубном пространстве
подогревателя питательной воды 3, в
трубном пространстве выносного
теплообменника 4 до
65°С
за счет холодного циркулирующего газа,
идущего по межтрубному пространству,
и затем в аппаратах воздушного охлаждения
7
до 40°С,
при этом часть аммиака конденсируется.
Жидкий аммиак, сконденсировавшийся при
охлаждении, отделяется в сепараторе
6,
а затем смесь, содержащая 10-12% NH3,
идет на циркуляционное колесо компрессора
5
азотоводородной смеси, где сжимается
до 32 МПа. Циркуляционный газ при
температуре 50°С
поступает в систему вторичной конденсации,
включающую конденсационную
колонну 8
и испарители
жидкого аммиака 15.
В конденсационной колонне газ охлаждается
до 18°С
и в испарителе за счет кипения аммиака
в межтрубном пространстве до —
5°С.
Из трубного пространства испарителей
смесь охлажденного циркуляционного
газа и сконденсировавшегося аммиака
поступает в сепарационную часть
конденсационной колонны, где происходит
отделение
жидкого аммиака
от газа
и смешение свежей азотоводородной смеси
с циркуляционным газом. Далее газовая
смесь проходит корзину с фарфоровыми
кольцами Рашига, где отделяется от
капель жидкого аммиака, поднимается по
трубкам теплообменника и направляется
в выносной теплообменник 4, а затем в
колонну синтеза.
Жидкий аммиак из первичного сепаратора проходит магнитный фильтр 16, где из него выделяется катализаторная пыль, и смешивается с жидким аммиаком из конденсационной колонны 8. Затем его дросселируют до давления 4Мпа и отводят в сборник жидкого аммиака 11. В результате дросселирования жидкого аммиака до 4Мпа происходит выделение растворенных в нем газов Н2, N2, O2, CH4. Эти газы, называемые танковыми, содержат 16-18% NН3. Поэтому танковые газы направляют в испаритель 12 с целью утилизации аммиака путем его конденсации при — 25°С. Из испарителя танковые газы и сконденсировавшийся аммиак поступает в сепаратор 13 для отделения жидкого аммиака, направляемого в сборник жидкого аммиака 11.
Для поддержания в циркуляционном газе постоянного содержания инертных газов, не превышающего 10%, производится продувка газа после первичной конденсации аммиака (после сепаратора 6). Продувочные газы содержат 8-9% NН3, который выделяется при температуре — 25-30°С в конденсационной колонне 9 и испарители 10 продувочных газов. Смесь танковых и продувочных
Резюме
• Формально побочных продуктов в этом простом по стехиометрии процессе нет, но отдувка части циркулирующего газа необходима для удаления примесей, от которых при очистке не удаётся избавиться полностью. Кроме того, имеются танковые газы, которые выделяются из жидкого аммиака после снижения давления. Из этих газов выделяют аммиак в испарителях 10 и 12 при температуре -25 - -30о и используют в качестве топливного газа.
• Т.о. производство аммиака представляет малоотходный процесс, в котором реализуются все принципы химической технологии.
5.3. Производство азотной кислоты. Основные стадии производства азотной кислоты. Окисление аммиака. Механизм, равновесие и кинетика процесса окисления аммиака. Стадии гетерогенно-каталитического процесса на платиновом катализаторе. Обоснование выбора оптимальных условий процесса: катализатора, температуры, давления, соотношения реагентов, времени контактирования. Окисление оксида азота(II). Особенности кинетики процесса и обоснование выбора оптимальных условий проведения процесса. Абсорбция диоксида азота. Уравнение для скорости абсорбции оксида азота(IV). Выбор оптимальных условий процесса: абсорбента, температуры, направления движения потоков. Очистка отходящих газов от оксидов азота. Энерготехнологическая схема производства азотной кислоты под давлением 0,716 МПа. Реализация технологических принципов в производстве азотной кислоты.
Процесс производства разбавленной азотной кислоты складывается из трех стадий:
1) конверсии аммиака с целью получения оксида азота
4NH3 + 5О2 = 4NO + 6Н2О; (IX)
2) окисления оксида азота до диоксида азота
2NO + О2 = 2NО2;
3) абсорбции оксидов азота водой
3NО2 + Н2О = 2HNО3 + NO
Суммарная реакция образования азотной кислоты выражается уравнением
Побочные:
4NH3 + 3O2 = 2N2 + 6H2O
4NH3 + 4O2 = 2N2O + 6H2O
2NH3 = N2 + 3H2
2NO = N2 + O2
4NH3 + 6NO = 5N2 + 6H2O
∆Н = - 328 кДж/моль
∆Н = - 1156 кДж/моль
∆Н = + 91 кДж/моль
∆Н = -180 кДж/моль
∆Н = -1810 кДж/моль
Каталитическое окисление аммиака - многостадийный гетерогенно-каталитический процесс, протекающий во внешнедиффузионной области и лимитируемый диффузией аммиака к поверхности катализатора. Предположениям об образовании в процессе окисления NH3 нестойких промежуточных соединений, которые в результате распада и перегруппировки дают оксид азота (II) и элементарный азот.
Скорость каталитического окисления аммиака по реакции (IX) очень высока. За десятитысячные доли секунды степень превращения аммиака в оксид азота (11) достигает 97-98% при атмосферном давлении и 95-96% под давлением до 0,88-0,98 МПа. Однако выход оксида азота (II) может быть различным на одном и том же катализаторе в зависимости от выбранных технологических параметров: температуры, давления, линейной скорости газа, содержания аммиака в аммиачно-воздушной смеси, напряженности катали затора, числа сеток и некоторых других факторов.
Катализаторы:
Катали заторы, применяемые для окисления аммиака, должны обладать избирательными свойствами, т. с. ускорять только одну реакцию (IX) окисления аммиака до оксида азота (II). Наиболее селективным и активным катализатором данной реакции оказался платиноидный катализатор, представляющий собой сплав платины с палладием и родием. В промышленности чаше всего применяют платино-родиевые сплавы (кроме платины до 10% родия, палладия, рутения, осмия и др.) в виде сеток.
Окисление аммиака на этом катализаторе протекает очень быстро, при времени контактирования порядка 10-4 с, а выход NO составляет примерно 98%.
Неплатиновые катализаторы, состоящие из оксидов железа и хрома, дают выход оксида азота (II) около 96%, но скорость реакции примерно в 100 раз меньше.
Скорость гетерогенного каталитического процесса зависит от величины поверхности катализатора. Для увеличения поверхности и сокращения диффузионного пути молекул платиновый катализатор изготавливают в виде сеток из тончайших нитей диаметром 0,1 мм.
Влияние температуры. Температура оказывает наибольшее влияние на выход оксида азота (II). При повышении температуры выход NO возрастает (рис. 13.1 8), причем существует оптимальная температура (для чистой платины 900-920 Т), при которой достигается максимальный выход оксида азота.

Н а
платине реакция окисления аммиака
начинается при 195°C. Особенностью
окисления
является то, что сначала происходит
так называемое мягкое окисление аммиака
до молекулярного
азота.
Заметное количество оксида азота (II)
начинает появляться при 300°С. С повышением
температуры выход оксида азота (II)
растет, достигая максимального значения
96% на чистой платине и 99% на сплавах
Pt-Pd-Rh.
Проведение процесса при высоких
температурах помимо увеличения выхода
оксида азота (II) имеет и другие
преимущества: растет
скорость
реакции окисления аммиака и уменьшается
время контактирования.
Так, при повышении температуры с 650 до
900°С время контактирования сокращается
с 5 · I0-4
до 1,1 · I0-4
с. Но с повышением температуры
увеличиваются потери
дорогостоящей платины,
т. е. ухудшаются экономические показатели
процесса.
а
платине реакция окисления аммиака
начинается при 195°C. Особенностью
окисления
является то, что сначала происходит
так называемое мягкое окисление аммиака
до молекулярного
азота.
Заметное количество оксида азота (II)
начинает появляться при 300°С. С повышением
температуры выход оксида азота (II)
растет, достигая максимального значения
96% на чистой платине и 99% на сплавах
Pt-Pd-Rh.
Проведение процесса при высоких
температурах помимо увеличения выхода
оксида азота (II) имеет и другие
преимущества: растет
скорость
реакции окисления аммиака и уменьшается
время контактирования.
Так, при повышении температуры с 650 до
900°С время контактирования сокращается
с 5 · I0-4
до 1,1 · I0-4
с. Но с повышением температуры
увеличиваются потери
дорогостоящей платины,
т. е. ухудшаются экономические показатели
процесса.
Повышение температуры с 780 до 850°С приводит к увеличению прямых потерь катализатора почти вдвое.
При выборе температуры конверсии необходимо также учитывать наличие примесей в аммиачно-воздушной смеси. Температура должна быть тем выше, чем больше примесей содержится в исходной газовой смеси.
Влияние давления. С ростом давления наблюдается снижение выхода оксида азота (II). Однако использован невысокого давления при окислении аммиака позволяет повысить производительность агрегата, уменьшить размеры аппаратов. На современных крупных агрегатах производства азотной кислоты процесс окисления аммиака осуществляется под давлением 0,41-0,73 МПа.
Основным условием получения высоких выходов NO под давлением выше атмосферного являются повышение температуры и времени контактирования (увеличение числа стенок). Из рис. 13.19 видно, что для обеспечения выхода оксида азота (II) более 98% при давлениях 0,41-0,71 М Па необходимы температуры выше 950 С.
Повышением давления в процессе конверсии можно увеличить линейную скорость газа и напряженность катализатора, что в свою очередь связано с ростом числа катализаторных сеток. Из рис. 13.20 видно, что увеличение числа сеток приведет к повышению степени конверсии аммиака и линейной скорости газа.
Влияние концентрации аммиака. В промышленности объемное отношение поддерживается в пределах 1,7-1,9 для платиновых катализаторов и 2,1-2,2 – для оксидных. Для получения высокого выхода NO необходим примерно 30%-ный избыток кислорода сверх стехиометрического. Это связано с тем, что поверхность платинового катализатора должна быть постоянно покрыта кислородом (в отсутствие кислорода аммиак уже при 500°С начинает разлагаться на азот и водород).

Окисление оксида азота (II).
Нитрозные газы, полученные при окислении аммиака, содержат оксид азота (II), азот, кислород и пары воды. При переработке нитрозных газов в азотную кислоту необходимо окислить оксид азота (II) до диоксида. Реакция окисления
2NO + О2 ↔ 2NО2; ∆Н = -124 кДж
обратима, протекает с уменьшением объема и сопровождается выделением теплоты. Следовательно, в соответствии с принципом Ле Шателье снижение температуры и повышение давления способствуют смещению равновесия реакции вправо, т. е. в сторону образования NО2.
При температуре до 100°С равновесие реакции полностью сдвинуто в сторону образования NО2. При температуре выше 700°С образования диоксида азота не происходит. В связи с этим в горячих нитрозных газах, выходящих из контактного аппарата, NО2 отсутствует, и для его получения газовую смесь необходимо охладить.
Применение в производстве азотной кислоты воздуха, обогащенного кислородом, или чистого кислорода позволяет получать нитрозные газы с повышенным содержанием оксида азота (1 1) и увеличить скорость окисления NO в NО2.
Реакция окисления NO в NО2 ускоряется при понижении температуры, а с повышением температуры замедлятся почти до полного прекращения. Одна из гипотез заключается в том, что окисление NO в NО2 идет через образование промежуточного продукта- димера оксида азота (11):
2NO ↔ (NО)2 ∆Н < О
О2 + (NО)2 ↔ 2 NО2 ∆Н <О.
В установках, работающих под атмосферным давлением, окисляют оксид азота примерно на 92%, а оставшийся NO поглощают (совместно с NО2) щелочью, так как для окисления понадобилось бы много времени и соответственно большие объемы аппаратуры. Обычно переработку нитрозных газов в разбавленную кислоту проводят при температурах 10-50°С.
Абсорбция нитрозных газов.
Химическое взаимодействие NO2 протекает по следующим реакциям:
2NO2 + H2O = HNO3 + HNO2
3HNO2 = HNO3 + 2NO + H2O
∆Н = - 126 кДж (3.10)
∆Н = + 76 кДж (3.11)
Суммарное взаимодействие NO2 c водой можно представить уравнением реакции:
3NO2 + H2O = 2HNO3 + NO ∆Н = - 136 кДж (3.12.)
Все оксиды азота, за исключением NO, взаимодействуют с водой с образованием азотной кислоты. Поглощение оксидов азота водой связано с растворением в ней NO2, N2O4, N2O3 и образованием азотной и азотистой кислот. Азотистая кислота является малоустойчивым соединением и распадается на азотную кислоту, оксид азота (II) и воду.
Механизм образования разбавленной азотной кислоты можно представить так. В газовой фазе NO2 и N2O4 постоянно находятся в состоянии химического равновесия и их перенос на поверхность соприкасающихся фаз совершается в соответствии с законами молекулярной диффузии газов. В пограничном слое газ-жидкость NO2 переходит в жидкую фазу. Затем после растворения NO2 происходит химическая реакция (3.10), которая по сравнению с процессом диффузии протекает относительно быстро. Далее в жидкой фазе происходит сравнительно медленное разложение азотистой кислоты по реакции (3.11). Образующийся NO частично окисляется в растворе кислородом, но его большая часть взаимодействует с кислородом уже в газовой фазе по реакции (2NO + О2 ↔ 2NO2). Одновременно с абсорбцией и протеканием химических реакций в растворе в газовой фазе частично происходят те же реакции, приводящие к образованию азотной кислоты.
Медленным процессом, определяющим скорость поглощения оксидов азота, является диффузия их в жидкую фазу. При взаимодействии паров воды и диоксида азота в газовой фазе происходит образование кислотного тумана, вследствие этого создается дополнительное сопротивление при поглощении оксидов азота.
С понижением температуры и концентрации кислоты и повышением давления степень превращения диоксида азота растет. При концентрации азотной кислоты выше 65% поглощение почти прекращается.
Абсорбент |
Вода |
Температура |
10 – 40°С |
Направление потоков |
противоток |
Концентрация получаемой азотной кислоты |
53 - 56 % |
Технологическая схема производства азотной кислоты под давлением 0,716 МПа
О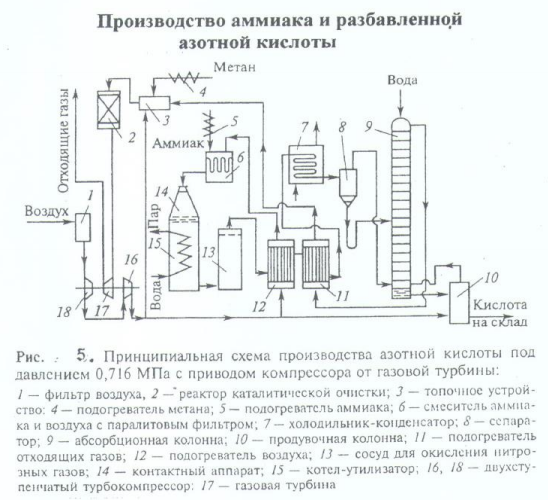 писание
технологической схемы производства
азотной кислоты.
писание
технологической схемы производства
азотной кислоты.
Атмосферный воздух проходит очистку в фильтре 1. Очищенный воздух сжимают двухступенчатым воздушным компрессором (16,18). Основной поток воздуха после сжатия нагревают в подогревателе воздуха 12 до 250-270°С теплотой нитрозных газов и подают на смешение с аммиаком в смеситель 6.
Газообразный аммиак, после очистки от влаги, масла и катализаторной пыли через подогреватель 5 при температуре 150 °С также направляют в смеситель 6. После очистки аммиачно-воздушную смесь с содержанием NH3 не более 10% подают в контактный аппарат 14 на конверсию аммиака.
К онверсия
аммиака протекает на платинородиевых
сетках (4) при температурах 870-900
ºС,
причем степень конверсии составляет
≈ 96%. Нитрозные газы при 890-910°С
поступают в котел-утилизатор
15,
расположенный под контактным аппаратом.
В котле за счет охлаждения
нитрозных газов до 170 °С
происходит испарение
воды,
питающей котел-утилизатор; при этом
получают пар с давлением 1,5
МПа и температурой 230°С.
онверсия
аммиака протекает на платинородиевых
сетках (4) при температурах 870-900
ºС,
причем степень конверсии составляет
≈ 96%. Нитрозные газы при 890-910°С
поступают в котел-утилизатор
15,
расположенный под контактным аппаратом.
В котле за счет охлаждения
нитрозных газов до 170 °С
происходит испарение
воды,
питающей котел-утилизатор; при этом
получают пар с давлением 1,5
МПа и температурой 230°С.
Нитрозные газы поступают в окислитель нитрозных газов 13. Частично окисление нитрозных газов происходит уже в котле-утилизаторе (до 40%). В окислителе 13 степень окисления возрастает до 85%. За счет реакции окисления нитрозные газы нагреваются до 300-335°С. Эта теплота используется в подогревателе воздуха 12.
Охлажденные в теплообменнике 12 нитрозные газы поступают в теплообменник 11, где происходит снижение их температуры до 150 °С и нагрев выхлопных (хвостовых) газов до 110-125°С. Затем нитрозные газы направляют в холодильник-конденсатор 7. При этом конденсируются водяные пары, и образуется слабая азотная кислота. Нитрозные газы отделяют от сконденсировавшейся азотной кислоты в сепараторе 8, из которого азотную кислоту направляют в адсорбционную колонну 9 на 6-7-ю тарелку, а нитрозные газы - под нижнюю тарелку абсорбционной колонны. Кислота направляется в продувочную колонну 10, а отбеленная азотная кислота поступает на склад. Воздух после продувочной колонны подается в нижнюю часть абсорбционной колонны 9.
Степень абсорбции оксидов азота достигает 99%. Выходящие из колонны хвостовые газы с содержанием оксидов азота до 0,11% при температуре 35°С проходят подогреватель 11, где нагреваются до 110-145°С и поступают в топочное устройство (камера сжигания 3 установки каталитической очистки). Здесь газы нагреваются до температуры 390-450°С за счет горения природного газа, подогретого в подогревателе 4, и направляются в реактор с двухслойным катализатором (палладий на оксиде алюминия) 2. Очистку осуществляют при 760°С. Очищенные газы поступают в газовую турбину 17 при температуре 690-700°С; энергия, вырабатываемая турбиной за счет теплоты хвостовых газов, используется для привода турбокомпрессора 18.
Содержание оксидов азота в очищенных выхлопных газах составляет 0,005-0,008 %, содержание СО2 - 0,23%.
Данный агрегат по производству 53-58 % -ной азотной кислоты мощностью 120 тыс.т/год полностью автономен по энергии. Энергия рекуперируется в результате установки на одной оси с турбокомпрессором газовой турбины.





5.4. Производство серной кислоты. Структура сырьевой базы. Получение оксида серы (IV) окислением серосодержащего сырья. Выбор условий процесса. Контактное окисление оксида серы (IV) в оксид серы (VI). Равновесие и кинетика процесса. Катализаторы. Выбор оптимальных условий процесса: катализатора, температуры, давления, соотношения исходных реагентов. Линия оптимальных температур. Абсорбция оксида серы (VI). Уравнение для скорости абсорбции оксида серы(VI). Обоснование выбора условий ее проведения: абсорбента, температуры, направления потоков. Технологическая схема получения серной кислоты из серы по методу ДК-ДА. Реализация технологических принципов в производстве серной кислоты.
Производство серной кислоты состоит из 3 стадий:
1. Получение SO2 .
2. Окисление SO2 в SO3 .
3. Абсорбция SO3 .
Сырьё:
1. Самородная сера.
2 .
Сульфиды металлов MS2
(FeS2)
.
Сульфиды металлов MS2
(FeS2)
3. Газовая сера. (преобладающее сырьё)
Газовая сера получается:
1)вначале производится гидрирование сероорганических соединений, содержащихся в попутных нефтяных газах, природном газе до H2S
2H2S + 3O2 = 2SO2 + 2H2O T= 900-1350оС
2H2S + SO2 = 3S + 2H2O Al2O3 , Т = 220-250оС
Сырье для серной кислоты и методы ее получения. Исходными реагентами для получения серной кислоты могут быть, элементная сера и серосодержащие соединения, из которых можно получить либо серу, либо диоксид серы. Такими соединениями являются сульфиды железа, сульфиды цветных металлов (меди, цинка и др.), сероводород и ряд других сернистых соединений.
Исходное сырье:
1. самородная сера;
2. газовая сера;
3. железный (серный) колчедан – FeS2;
4. отходящие газы цветной металлургии;
5. газы переработки сульфатов металлов.
Традиционно основные источники сырья - сера и железный (серный) колчедан. Постепенно доля колчедана как сырьевого источника уменьшается, что связано и с большими транспортными расходами на его транспортировку (кроме серы в нем весьма велика доля других компонентов), и невозможностью избавиться от отхода - огарка. Значительное место в сырьевом балансе производства серной кислоты занимают отходящие газы цветной металлургии, содержащие диоксид серы.
Для защиты окружающей среды во всем мире принимаются меры по использованию отходов промышленности, содержащих серу. В атмосферу с отходящими газами тепловых электростанций и металлургических заводов выбрасывается диоксида серы значительно больше, чем применяется для производства серной кислоты. В то же время из-за низкой концентрации SО2 в таких отходящих газах их переработка пока еще не всегда осуществима.
1. Контактный метод получения серной кислоты. Рассмотрим процесс получения серной кислоты контактным методом из двух видов сырья: серного (железного) колчедана и серы. Циклонная печь для сжигания серы состоит из двух горизонтальных цилиндров- форкамеры 1 и двух камер дожигания 2 и 3. В форкамеру через две группы сопл 9 подают воздух, через форсунку механического типа 8 поступает расплавленная сера. Образующейся при сжигании жидкой серы обжиговый газ вместе с парами серы поступает через пережимное кольцо 2 из форкамеры в первую камеру дожигания, в которой также расположены воздушные сопла 11 и форсунки для подачи серы 10. Из первой камеры дожигания газ через пережимное кольцо 4 поступает во вторую камеру дожигания 3, где сгорают остатки серы (в пространстве между пережимными кольцами 4 к газу добавляют воздух). Первой стадией процесса является окисление сырья с получением обжигового газа, содержащего диоксид серы. В зависимости от вида сырья протекают экзотермические химические реакции обжига:
4FeS2 + 11O2 = 2Fe2O3 + 8SO2; ΔH = - 3415 кДж (1)
S + O2 = SO2 (2)
П
 ри
протекании реакции (1) помимо газообразного
продукта реакции SO2
образуется твердый продукт Fe2O3,
который может
присутствовать
в газовой фазе в
виде пыли.
Колчедан содержит
различные примеси,
в частности соединения мышьяка
и фтора,
которые в процессе обжига переходят
в газовую фазу.
Присутствие этих соединений на стадии
контактного окисления диоксида серы
может
вызвать отравление катализатора.
Поэтому реакционный газ
после стадии обжига колчедана должен
быть
предварительно направлен
на стадию подготовки
к
контактному окислению (вторая стадия),
на которой помимо очистки
от
каталитических ядов,
выделяются
пары воды
(осушение), а также получаются
побочные продукты
(Se и Те).
ри
протекании реакции (1) помимо газообразного
продукта реакции SO2
образуется твердый продукт Fe2O3,
который может
присутствовать
в газовой фазе в
виде пыли.
Колчедан содержит
различные примеси,
в частности соединения мышьяка
и фтора,
которые в процессе обжига переходят
в газовую фазу.
Присутствие этих соединений на стадии
контактного окисления диоксида серы
может
вызвать отравление катализатора.
Поэтому реакционный газ
после стадии обжига колчедана должен
быть
предварительно направлен
на стадию подготовки
к
контактному окислению (вторая стадия),
на которой помимо очистки
от
каталитических ядов,
выделяются
пары воды
(осушение), а также получаются
побочные продукты
(Se и Те).
Если обжиговый газ получают сжиганием серы, то отпадает необходимость очистки от примесей. Стадия подготовки будет включать в себя лишь осушку газа и утилизацию теплоты.
На третьей стадии протекает обратимая экзотермическая химическая реакция контактного окисления диоксида серы:
SO2 +1/2 O2 ↔SO3 ΔH < 0;
Последняя стадия процесса - абсорбция триоксида серы концентрированной серной кислотой или олеумом.
Важнейшей задачей в производстве серной кислоты является повышение степени превращения SО2 в SО3. Помимо увеличения производительности по получению серной кислоты выполнение этой задачи позволяет решить и экологические проблемы – снизить выбросы в окружающую среду вредного компонента SО2.
П овышение
степени превращения
SО2
может быть достигнуто созданием схемы
двойного
контактирования
и двойной
абсорбции
(ДКДА).
Другим
возможным вариантом решения той же
задачи является проведение процесса
по циклической (замкнутой) схеме с
применением технического
кислорода.
овышение
степени превращения
SО2
может быть достигнуто созданием схемы
двойного
контактирования
и двойной
абсорбции
(ДКДА).
Другим
возможным вариантом решения той же
задачи является проведение процесса
по циклической (замкнутой) схеме с
применением технического
кислорода.
Получение обжигового газа из серы. При сжигании серы протекает необратимая экзотермическая реакция (2) с выделением очень большого количества теплоты ΔH = -362,4 кДж/моль. Расплавленная жидкая сера, подаваемая на сжигание, испаряется (кипит) при температуре 444,6°C. Теплоты реакции горения серы вполне достаточно для испарения исходного сырья, поэтому взаимодействие серы и кислорода происходит в газовой фазе (гомогенная реакция).
Сжигание серы в промышленности. Серу предварительно расплавляют (для этого можно использовать водяной пар, полученный при утилизации теплоты основной реакции горения серы). Так как температура плавления серы сравнительно низка, то отстаиванием и последующей фильтрацией от серы легко отделить механические примеси, не перешедшие в жидкую фазу, и получить исходное сырье достаточной степени чистоты. Для сжигания расплавленной серы используют два типа печей – форсуночные и циклонные. В них необходимо предусмотреть распыление жидкой серы для ее быстрого испарения и обеспечить надежный контакт с воздухом во всех частях аппарата.
Концентрация диоксида серы в обжиговом газе зависит от соотношения серы и воздуха, подаваемых на сжигание. Если воздух берут в стехиометрическом количестве, т. е. на каждый моль серы 1 моль кислорода, то при полном сгорании серы концентрация будет равна объемной доле кислорода в воздухе CSO2max = 21%.
Однако обычно воздух берут в избытке, так как в противном случае в печи будет слишком высокая температура. При адиабатическом сжигании серы температура обжига для реакционной смеси стехиометрического состава составит примерно 1500°С. В практических условиях возможности повышения температуры в печи ограничены тем, что выше 1300°C быстро разрушается футеровка (облицовка) печи и газоходов. Обычно при сжигании серы получают обжиговый газ, содержащий 13-14% SО2.
Получение обжигового газа из колчедана. Суммарную реакцию обжига колчедана можно представить в виде реакции (1). Она протекает необратимо, сильно экзотермическая и состоит из трех стадий:
1. FeS2 = FeS + S; ΔH > 0;
2. 4FeS + O2 = 2Fe2O3 + 4SO2; ΔH < 0;
3. S + O2 = SO2; ΔH < 0;
Часть кислорода воздуха расходуется в реакции на окисление железа и поэтому максимально возможная концентрация диоксида серы в обжиговом газе в этом случае ниже, чем при сжигании серы.
В состав обжигового газа входит также небольшое количество триоксида серы SО3, так как оксид железа при высоких температурах является катализатором окисления SО2 в SО3.
Обжиг колчедана - типичный гетерогенный процесс в системе «газ-твердое», который можно описать моделью с фронтальным перемещением зоны реакции. В соответствии с этой моделью процесс включает в себя ряд диффузионных стадий и саму химическую реакцию, также многостадийную. Для увеличения скорости процесса стремятся прежде всего уменьшить сопротивление диффузионных стадий, т. е. не проводить обжиг колчедана в диффузионной области. Это может быть достигнуто измельчением твердой фазы и интенсивной турбулизацией потока. Наиболее удобным аппаратом для этой цели является печь с псевдоожиженным слоем колчедана (печь «кипящего слоя» КС).
Температура процесса должна быть достаточно большой для обеспечения высокой скорости реакции. При низких температурах (ниже 500°C) не сможет протекать эндотермическая реакция термического разложения дисульфида железа. Однако проведение обжига при очень высоких температурах может вызвать нежелательный физический процесс спекания частиц горячего материала, приводящий к увеличению их размеров. Следствием этого явится возрастание времени полного превращения твердых частиц и понижение производительности печи. Температура спекание колеблется в зависимости от состава (сорта) колчедана от 800 до 900°С.
Проведение процесса в адиабатическом режиме привело бы к разогреву до более высоких температур. Поэтому часть теплоты обжига приходится отводить внутри печи. Удобнее всего это сделать в печах КС, так как в псевдоожиженном слое твердого материала достаточно велик коэффициент теплоотдачи от колчедана к поверхности охлаждающих элементов и в «кипящий» слой можно ввести змеевики охлаждения.
При производстве серной кислоты для обжига колчедана применяют в основном печи кипящего слоя с псевдоожиженным слоем твердого материала. В псевдоожиженном слое обеспечивается высокая скорость диффузионных и теплообменных процессов (подвод кислорода к поверхности колчедана, отвод диоксида серы в газовый поток, отвод теплоты от поверхности сырья к газовому потоку). Отсутствие тормозящего влияния масса- и теплообмена позволяет проводить обжиг колчедана в таких печах с высокой скоростью. Печи КС характеризуются максимальной интенсивностью в сравнении с другими конструкциями, применяемыми для обжига колчедана. К недостаткам печей КС можно отнести высокую запыленность обжигового газа.
Подготовка обжигового газа к контактному окислению. Из обжигового газа необходимо удалить примеси, а также нагреть (или охладить) их до температуры, при которой начинается контактное окисление. Обжиговый газ, полученный сжиганием колчедана в печах КС, содержит большое количество огарковой пыли, соединения мышьяка, селена и фтора. Очистка обжигового газа начинается в печном отделении, где в циклонах и сухих электрофильтрах осаждают огарковую пыль. Содержание пыли в газе после этих аппаратов сухой очистки не должно превышать 50 мг/м3.
Наличие в газе пыли, даже в небольших количествах оставшейся после сухой очистки, может привести к повышению гидравлического сопротивления аппаратов и отравлению катализатора соединениями As, адсорбированными на огаркавой пыли.
Затем газ направляют на стадию мокрой очистки (в промывное отделение), где из обжигового газа удаляют остатки пыли, каталитические яды (соединения мышьяка и фтора), а также соединения селена. Мокрая очистка обжигового газа заключается в промывке его разбавленной серной кислотой. При этом происходит ряд физических процессов: конденсация, абсорбция и т. п. Основные примеси обжигового газа, находящиеся в газо- и парообразном состоянии, выделяются при промывке серной кислотой, имеющей более низкую температуру, чем очищаемый газ. Примеси частично растворяются в серной кислоте, но большая их часть переходит в состав сернокислотного тумана.
Суммарная поверхность капель тумана серной кислоты весьма велика, поэтому в них растворяется большое количество примесей, выделяющихся из газа вместе с туманом в промывных башнях и электрофильтрах. Тщательная очистка газа от тумана необходима для выделения не только примесей, отравляющих контактную массу, но и содержащейся в каплях серной кислоты, иначе при прохождении газа через аппаратуру и трубопровод ы будет происходить коррозия.
Подготовка к контактному окислению газа, полученного при сжигании серы, значительно проще. Сера не содержит примесей, которые при ее сжигании могли бы стать каталитическими ядами. Поэтому очистка газа заключается лишь в его осушке. Так как осушка концентрированной серной кислотой происходит при низких температурах, целесообразно подвергать осушке не обжиговый газ, который пришлось бы специально охлаждать, а холодный воздух, подаваемый на сжигание серы. Обжиговый газ в этом случае будет содержать лишь минимальное (допустимое) количество паров воды и для проведения контактного окисления его нужно лишь охладить в котлах-утилизаторах до температуры зажигания катал и затора.
Контактное окисление диоксида серы. Реакция (III) окисления диоксида серы характеризуется очень высоким значением энергии активации и поэтому практическое се осуществление возможно лишь в присутствии катализатора.
В промышленности основным катализатором окисления SO2 является катализатор на основе пентоксида ванадия V2О5, (ванадиевая контактная масса). Каталитическую активность в этой реакции проявляют и другие соединения. прежде всего платина. Однако платиновые катализаторы чрезвычайно чувствительны даже к следам мышьяка, селена, хлора и других примесей и поэтому постепенно были вытеснены ванадиевыми катализаторами.
Каталитическую активность проявляет также оксид железа (III) Fe2О3, однако лишь в области высоких температур. Каталитической активностью Fe2О3, входящего в состав огарка, можно объяснить наличие в обжиговом газе, выходящем из печей КС, небольших количеств триоксида серы.
Скорость реакции повышается с ростом концентрации кислорода, поэтому процесс в промышленности проводят при его избытке. Например, при получении серной кислоты из колчедана состав газа, подаваемого на контактное окисление, поддерживают таким (в объемных долях, %): SO2 - 7-9; O2 - 9-11; N2 - 82. Таким образом, для осуществления реакции с высокой скоростью кислород берут почти в трехкратном избытке по отношению к стехиометрическому количеству. Для этого более концентрированный обжиговый газ (14-15% SO2) разбавляют воздухом перед стадией контактного окисления.
Так как реакция окисления SO2 относится к типу экзотермических, температурный режим ее проведения должен приближаться к линии оптимальных температур. На выбор температурного режима дополнительно накладываются два ограничения, связанные со свойствами катализатора. Нижней предельной является температура зажигания ванадиевых катализаторов, составляющая в зависимости от конкретного вида катализатора и состава газа 400-440°С. Верхняя предельная температура составляет 600-650°С. Выше этих температур происходит перестройка структуры катализатора и он теряет свою активность.
В диапазоне температур 400-600°C процесс стремятся провести так, чтобы по мере увеличения степени превращения температура уменьшалась.
Один из наиболее рациональных методов, повсеместно применяемый при производстве серной кислоты - метод двойного контактирования и двойной абсорбции (ДКДА). Его сущность состоит в том, что реакционную смесь, в которой степень превращения SO2 составляет 90-95%, охлаждают и направляют в промежуточный абсорбер для выделения SO3. В оставшемся реакционном газе отношение O2:SO2 существенно повышается, что приводит к смещению равновесия реакции вправо. Вновь нагретый реакционный газ снова подают в контактный аппарат, где на одном-двух слоях катализатора достигают 95%-ной степени превращения оставшегося SO2. Суммарная степень превращения SO2 составляет в таком процессе 99,5-99,8%. При контактном отделении по методу ДКДА на первой стадии контактирования используются три слоя, на второй - один.
Абсорбция триоксида серы. Послед ней стадией процесса производства серной кислоты контактным способом является абсорбция триоксида серы из газовой смеси и превращение его в серную кислоту. При выборе абсорбента и условий проведения стадии абсорбции необходимо обеспечить почти 100%-е извлечение SO3 из газовой фазы.
Для полного извлечения SO3 необходимо, чтобы его равновесное парциальное давление над растворителем было ничтожно малым, так как при этом будет велика движущая сила процесса абсорбции. Однако в качестве абсорбента нельзя использовать и такие растворы, над поверхностью которых велико равновесное парциальное давление паров воды. В этом случае еще не растворенные молекулы SO3 будут реагировать с молекулами воды в газовой фазе с образованием паров серной кислоты и быстро конденсироваться в объеме с образованием мельчайших капель серной кислоты, т.е. с образованием сернокислотного тумана:
SO3 + Н2О↔Н2SO4 ΔH < 0;
Реакция обратимая, экзотермическая.
Туман плохо улавливается в обычной абсорбционной аппаратуре и в основном уносится с отходящими газами в атмосферу, при этом загрязняется окружающая среда и возрастают потери серной кислоты. Оптимальным абсорбентом является 98,3% серная кислота (техническое название- моногидрат), соответствующая азеотропному составу. Над этой кислотой нет ни паров воды, ни паров SO3. Протекающий при этом процесс можно описать уравнением реакции
SO3 + n Н2SO4 + Н2О ↔ (n + l) Н2SO4.
Использование в качестве поглотителя менее концентрированной серной кислоты может привести к образованию сернокислотного тумана, а над 100%-ной серной кислотой или олеумом в паровой фазе довольно велико равновесное парциальное давление SO3, по этому он будет абсорбироваться не полностью.
Однако в процессе абсорбции SO3 происходит закрепление кислоты (повышение ее концентрации) и из-за экзотермичности реакции увеличивается температура. Для уменьшения тормозящего влияния этих явлений абсорбцию ведут так, чтобы концентрация Н2SO4 при однократном прохождении абсорбера повышалась лишь на 1-1,5%. Закрепившуюся серную кислоту разбавляют в сборнике до концентрации 98,3%, охлаждают в наружном холодильнике и вновь подают на абсорбцию, обеспечивая высокую кратность циркуляции.
Побочные реакции при обжиге пирита:
1. 2FeS2 + 7O2 = Fe2(SO4)3 + SO2
2. FeS2 + 3O2 = FeSO4 + SO2
3. SO2 + O2 = SO3 и т.п.
Процесс обжига колчедана – сложный, практически необратимый, экзотермический, гетерогенный, некаталитический.
Выбор оптимальных условий проведения процесса.
Катализатор.
Процесс окисления диоксида серы с заметной скоростью для различных катализаторов начинается при определенной температуре – температуре зажигания. Реакция ускоряется в присутствии платины (Е=70 кДж/моль) при температуре 2500С, оксида железа (III) ( Е=150 кДж/моль) при температуре 5500С, оксида ванадия (V) при температуре не ниже 4000С ( Е= 90 кДж/моль). Платиновый катализатор обладает наибольшей активностью, однако дорог и быстро отравляется ядами ( мышьяком, селеном, хлором). Оксид железа (III) – малоактивный катализатор. Ванадиевая контактная масса, например, марки БАВ имеет примерный состав: V2O5 * 0,5Al2O3 *2K2O*3BaO*2KCl*12SiO2 Активными компонентами ванадиевых катализаторов являются сульфо- и пиросульфованадаты калия, которые в условиях проведения реакции находятся в расплавленном состоянии на поверхности кремнеземистого носителя. Формы контактной массы – гранулы, кольца. Рабочий интервал температур 400-6500С. При температурах выше 6500С активность катализатора уменьшается из-за разрушения активного комплекса V2O5 * K2S2O7 до кристаллического пентаоксида ванадия, который катализатором не является. При температурах ниже 4000С возможно образование каталитически неактивного соединения – сульфата ванадила VOSO4 .
Температура. Температура выбирается в пределах работы катализатора. Для получения высокого выхода оксида серы (VI) необходима минимальная температура- 4000С, так как реакция обратимая и экзотермическая. Однако скорость процесса при этой температуре мала даже в присутствии катализатора. Выбор температурного режима, обеспечивающего высокую скорость обратимой экзотермической реакции, довольно сложен, так как изменение температуры различно сказывается на равновесном выходе продукта и на средней скорости процесса. Если вести процесс при постоянной температуре 6000С, то реакция идет быстро, но, в соответствии с состоянием равновесия, выход целевого продукта уменьшается. Если вести процесс при постоянной температуре 4000С, получим высокий выход, однако скорость процесса будет чрезвычайно мала. Для обеспечения высокой интенсивности процесса необходимо проводить окисление оксида серы (IV) при меняющемся температурном режиме.
Давление. Процесс идет с уменьшением объема, поэтому для смещения равновесия вправо необходимо давление увеличивать. В зависимости от выбранной температуры эффективность воздействия давления различна. При низких температурах, когда равновесные степени контактирования оксида серы (IV) высокие, давление незначительно сказывается на смещении равновесия. При высоких температурах, когда окисление происходит далеко не полностью, давление может стать одним из решающих факторов, обеспечивающих высокую степень контактирования. Повышенное давление имеет значение для агрегатов большой единичной мощности. В обычных условиях процесс проводят при давлении 0,1 МПа, так как смещения равновесия добиваются другими путями. Степень превращения SO2 составляет в среднем 99,5-99,8%.
Соотношение исходных компонентов. При стехиометрическом соотношении превращение протекает недостаточно полно. Для смещения равновесия в сторону продукта реакции необходимо брать в избытке один из компонентов. Наиболее оптимальным является следующий состав газовой смеси: 11%O2 , 7%SO2 , 82%N2 , т.е. кислород подают в избытке. Если взять избыток диоксида серы, то произойдет перегрев катализатора, что может вывести его из строя.
С еру
подают в печь 1, пропуская через плавитель,
где она плавится. Также в печь 1 подают
воздух, осушаемый в сушильной башне 9,
орошаемой 93% серной кислотой. Воздух
предварительно нагревается в
теплообменниках-8.2, 8.3. Из печи 1 газ (SO2
) поступает в котел-утилизатор 2, где
охлаждается до 4400С и направляется в
контактный аппарат 7. Воздух нагревается
в топке 4, в теплообменниках 5 и 6 и
направляется в контактный аппарат. В
контактном аппарате размещены пять
слоев катализатора. Для реализации
линии оптимальных температур газ после
каждого слоя катализатора необходимо
охладить. С этой целью предусмотрена
система теплообменников 8. Таким образом,
газовая смесь (SO2 и воздух) после первого
слоя катализатора направляется в
теплообменник 8.1, затем на второй слой
катализатора. После второго слоя газ
охлаждается в теплообменнике 8.4 и
поступает на третий слой катализатора.
После третьего слоя газ, пройдя
теплообменники 8.5, 8.2, 8.3, направляется
в первый моногидратный абсорбер 10,
который орошается 98,3% серной кислотой.
После первого моногидратного абсорбера
газ поступает на четвертый слой
катализатора в контактном аппарате,
предварительно пройдя теплообменники
8.5, 8.4 и 8.1. После четвертого слоя вводят
дополнительно воздух для охлаждения и
интенсификации процесса. После четвертого
слоя газ поступает на пятый слой, выходит
из контактного аппарата и направляется
для подогрева воды в экономайзер 3 и уже
оттуда попадает во второй моногидратный
абсорбер 11, где происходит окончательное
поглощение триоксида серы. Выхлопные
газы отводятся из второго моногидратного
абсорбера через выхлопную трубу 13.
Товарная серная кислота – 92,5% постоянно
выводится из сушильной башни 9.
еру
подают в печь 1, пропуская через плавитель,
где она плавится. Также в печь 1 подают
воздух, осушаемый в сушильной башне 9,
орошаемой 93% серной кислотой. Воздух
предварительно нагревается в
теплообменниках-8.2, 8.3. Из печи 1 газ (SO2
) поступает в котел-утилизатор 2, где
охлаждается до 4400С и направляется в
контактный аппарат 7. Воздух нагревается
в топке 4, в теплообменниках 5 и 6 и
направляется в контактный аппарат. В
контактном аппарате размещены пять
слоев катализатора. Для реализации
линии оптимальных температур газ после
каждого слоя катализатора необходимо
охладить. С этой целью предусмотрена
система теплообменников 8. Таким образом,
газовая смесь (SO2 и воздух) после первого
слоя катализатора направляется в
теплообменник 8.1, затем на второй слой
катализатора. После второго слоя газ
охлаждается в теплообменнике 8.4 и
поступает на третий слой катализатора.
После третьего слоя газ, пройдя
теплообменники 8.5, 8.2, 8.3, направляется
в первый моногидратный абсорбер 10,
который орошается 98,3% серной кислотой.
После первого моногидратного абсорбера
газ поступает на четвертый слой
катализатора в контактном аппарате,
предварительно пройдя теплообменники
8.5, 8.4 и 8.1. После четвертого слоя вводят
дополнительно воздух для охлаждения и
интенсификации процесса. После четвертого
слоя газ поступает на пятый слой, выходит
из контактного аппарата и направляется
для подогрева воды в экономайзер 3 и уже
оттуда попадает во второй моногидратный
абсорбер 11, где происходит окончательное
поглощение триоксида серы. Выхлопные
газы отводятся из второго моногидратного
абсорбера через выхлопную трубу 13.
Товарная серная кислота – 92,5% постоянно
выводится из сушильной башни 9.
5.5. Производство метанола. Получение синтез газа для производства метанола на основе пароуглекислотной конверсии метана. Химия, равновесие и кинетика процесса синтеза метанола. Высокотемпературные и низкотемпературные катализаторы. Обоснование выбора технологических параметров процесса: катализаторов, температуры, давления, соотношения исходных реагентов, объемной скорости процесса. Колонны синтеза метанола при высоком и низком давлениях. Технологическая схема производства метанола при высоком (32 МПа) и низком (5 МПа) давлениях. Реализация технологических принципов в производстве метанола.
Синтез метанола в промышленности в настоящее время проводится из синтез-газа, в состав которого входят водород, монооксид углерода и диоксид углерода. Основными реакциями синтеза метанола являются:
СО + 2Н2 ↔ СН3ОН (20) ΔН = - 90,8 кДж
СО2 + 3Н2 ↔ СН3ОН + Н2О (21) ΔН = - 49,6 кДж
Поэтому для конверсии природного газа с целью получения синтез-газа реализуют пароуглекислотную конверсию метана по реакциям (1) и (2):
СН4 + Н2О (пар) ↔ СО + 3Н2 (1)
СН4 + СО2 ↔ 2СО + 2Н2 (2)
В процессе синтеза метанола место ряд побочных реакций:
3) СО + 3Н2 ↔ СН4 + Н2О ΔН = - 115 кДж
4) 2СО + 4Н2 ↔ (СН3)2О+ Н2О ΔН = - 200 кДж
5) СН3ОН + nCO + 2nH2 ↔ СН3(CH2)nOH + n Н2О
6) 2СО ↔ СО2 + С
Синтез метанола: сложный, обратимый, экзотермический, с уменьшением числа молей.
Применяются два типа катализаторов:
Высокотемпературные катализаторы (цинк-хромовые катализаторы), на основе оксидов ZnO∙ Cr2O3∙Al2O3
Температурный интервал их работы 370- 420°С.
Низкотемпературные катализаторы (медьсодержащие) на основе оксидов СuO + Cr2O3 + Al2O3 + ZnO, причем медь восстанавливается водородом до металла. Температурный интервал 220-280°C. Необходимо отметить, что медьсодержащие катализаторы очень чувствительны к сернистым соединениям (катализаторные яды) и к перегреву.
На понижение температуры и уменьшение выхода метанола влияет введение в систему байпасного потока, который не только охлаждает реакционную смесь, но и разбавляет ее.
Синтез метанола проводят при повышенном давлении – при 5 или 30 МПа. При более высоких значениях давления начинают в большей степени идти побочные реакции, что резко снижает эффективность процесса.
При сравнительно низком давлении (5 МПа) используют низкотемпературные катализаторы, а при более высоком (32 МПа) высокотемпературные катализаторы.
Процесс проводят при небольшом избытке водорода по следующим причинами:
1. Для подавления побочных реакций –
СО + 3Н2 ↔ СН4 + Н2О (ΔН = - 115 кДж),
Реакции метанирования сильно экзотермичны, что может привести к перегреву катализатора и выходу его из строя, и, кроме того, ведут к ненужному в данном процессе метана.
2. Увеличивается скорость процесса синтеза, так как лимитирующей стадией процесса является хемосорбция водорода на катализаторе.
3. Увеличивается срок службы катализатора – избыток водорода предотвращает образование продуктов уплотнения.
4. Регулируется температура процесса, поскольку теплопроводность водорода выше, чем теплопроводность монооксида углерода.
Причины рециркуляции в синтезе метанола:
1. Наличие термодинамических ограничений (несмещаемость процесса)
2. Наличие избытка водорода
Т.к. в данном процессе есть рецикл исходных веществ, то надо осуществлять отдувку для вывода инертных примесей, которые как приходят с исходным сырьем, так и образуются с в процессе синтеза метанола (метан).
Катализатор |
Давление, МПа |
Температура, °С |
Соотношение H2/CO |
Объемная скорость, ч-1 |
Cu-содержащий |
5 |
220-280(по ЛОТ) |
|
15000-30000 |
Zn-содержащий |
32 |
370-390 |
Работают 2,1-2,2 |
|
Н еобходимо
отметить, что в данном случае в качестве
гидрирующего агента используется не
азотоводородная смесь (авс), а водород.
еобходимо
отметить, что в данном случае в качестве
гидрирующего агента используется не
азотоводородная смесь (авс), а водород.
Природный газ сжимается турбокомпрессором 1 до давления 3 МПа, подогревается в подогревателе 2 за счет сжигания в межтрубном пространстве природного газа и направляется на сероочистку в аппараты 3 и 4, где последовательно осуществляется каталитическое гидрирование органических соединений серы и поглощение образующегося сероводорода адсорбентом на основе оксида цинка. После этого газ смешивается с водяным паром и диоксидом углерода направляется в трубчатый конвертор 5, где на никелевом катализаторе происходит пароуглекислотная конверсия при температуре 850 - 870°С. Теплоту, необходимую для конверсии, получают в результате сжигания природного газа в специальных горелках, расположенных в межтрубном пространстве печи. Конвертированный газ поступает в котел-утилизатор 6, где охлаждается до 280-290°С. Затем теплоту газа используют в теплообменнике 7 для подогрева питательной воды, направляемой в котел-утилизатор. Пройдя воздушный холодильник 8 и сепаратор 9, в котором отделяют сконденсировавшуюся воду, газ охлаждается до 35 – 40°С. Охлажденный конвертированный газ сжимают до 5 МПа в компрессоре 10, смешивают с циркуляционным газом и подают в теплообменники 11, 12, где он нагревается до 220-230°С.
Нагретая газовая смесь поступает в колонну синтеза 13, температурный режим в которой регулируют с помощью холодных байпасов (проведение процесса синтеза метанола по ЛОТ). Далее газовая смесь охлаждается в холодильнике-конденсаторе 14, сконденсировавшийся метанол-сырец отделяется в сепараторе 15 и поступает в сборник 16. Циркуляционный газ возвращается на синтез, продувочные газы отдают на сжигание в трубчатую печь.
Вследствие снижения температуры синтеза при низком давлении процесс осуществляется в условиях, близких к равновесию, что позволяет увеличить производительность агрегата.


Схема при давлении в 32 МПа
Сжатый до 32 МПа синтез-газ очищается в масляном фильтре 1 и в угольном фильтре 2, после чего смешивается с циркуляционным газом и поступает в колонну синтеза 3.
Смешанный газ (на схеме – синтез-газ), пройдя через кольцевой зазор между катализаторной коробкой и корпусом колонны 3 (рис.11) (колонна синтеза метанола высокого давления – рис.13), поступает в межтрубное пространство теплообменника, расположенного в нижней части колонны (рис.13).
Смешанный газ, пройдя кольцевой зазор между катализаторной коробкой и корпусом колонны, поступает в межтрубное пространство теплообменника, расположенного в нижней части колонны. В теплообменнике газ нагревается до 330°С и по центральной трубе, в которой размещен электроподогреватель, поступает в верхнюю часть колонны и проходит последовательно пять слоев катализатора.
После каждого слоя катализатора, кроме последнего, в колонну вводят определенное количество холодного циркуляционного газа для поддержания необходимой температуры. После пятого слоя катализатора газ направляется в теплообменник, где охлаждается до 130°С и выходит из колонны. Размещение теплообменника внутри корпуса колонны, подача смешанного газа в колонну, значительно снижает потери тепла в окружающую среду (принцип наилучшего использования энергии).
Затем в холодильник-конденсатор типа «труба в трубе» 4 (рис.11). Здесь газ охлаждается до 30-35°С и продукты синтеза конденсируются.
М
 етанол-сырец
отделяют в сепараторе 5 и направляют в
сборник 7, после чего выводят на
ректификацию.
етанол-сырец
отделяют в сепараторе 5 и направляют в
сборник 7, после чего выводят на
ректификацию.
Г аз
проходит второй сепаратор 5 для выделения
капель метанола, компримируется до
давления синтеза турбо-циркуляционным
компрессором 6 и возвращается на синтез.
Продувочные газы выводят перед
компрессором и вместе с танковыми газами
используют в качестве топлива.
Технологический
процесс получения метанола из оксида
углерода и водорода включает ряд
операций, обязательных для любой
технологической схемы синтеза. Газ
предварительно очищается от карбонила
железа, сернистых соединений, подогревается
до температуры начала реакции и поступает
в реактор синтеза метанола. По выходе
из зоны катализа из газов выделяется
образовавшийся метанол, что достигается
охлаждением смеси, которая затем
сжимается до давления синтеза и
возвращается в процесс.
аз
проходит второй сепаратор 5 для выделения
капель метанола, компримируется до
давления синтеза турбо-циркуляционным
компрессором 6 и возвращается на синтез.
Продувочные газы выводят перед
компрессором и вместе с танковыми газами
используют в качестве топлива.
Технологический
процесс получения метанола из оксида
углерода и водорода включает ряд
операций, обязательных для любой
технологической схемы синтеза. Газ
предварительно очищается от карбонила
железа, сернистых соединений, подогревается
до температуры начала реакции и поступает
в реактор синтеза метанола. По выходе
из зоны катализа из газов выделяется
образовавшийся метанол, что достигается
охлаждением смеси, которая затем
сжимается до давления синтеза и
возвращается в процесс.
5 .6.
Производство
этанола.
Получение этанола методом сернокислотной
гидратации этилена. Производство этанола
прямой гидратацией этилена. Равновесие
и кинетика процесса. Обоснование условий
проведения процесса: катализатор,
температура, соотношение реагентов,
давление и объемная скорость.
Технологическая схема процесса прямой
гидратации этилена. Особенности
реализации технологических принципов
в производстве этанола.
.6.
Производство
этанола.
Получение этанола методом сернокислотной
гидратации этилена. Производство этанола
прямой гидратацией этилена. Равновесие
и кинетика процесса. Обоснование условий
проведения процесса: катализатор,
температура, соотношение реагентов,
давление и объемная скорость.
Технологическая схема процесса прямой
гидратации этилена. Особенности
реализации технологических принципов
в производстве этанола.




Температура процесса
Активность выбранного катализатора (83-85 %-ная фосфорная кислота) имеет решающее значение при установлении температурного режима работы реактора. Для промышленных условий это 280-300оС. При температуре ниже 2800С мала активность катализатора (небольшая скорость процесса), а выше 300оС- идут нежелательные процессы, которые приводят к падению селективности (полимеризация этилена, повышение скорости образования эфира и др.), поскольку энергия активации побочных реакций выше, чем для основной реакции образования этанола.
Соотношение реагентов Н2О/С2Н4 = 0,6-0,7/1 (Берется недостаток воды для уменьшения нагрузки на систему разделения (уменьшаются энергозатраты на процесс отделения этилового спирта от воды
Давление. Давление не должно превышать P= 7,5-8 МПа для предотвращения конденсации водяного пара. При значении РН2О > Р*Н2О (р* Н2О - равновесное давление паров воды над фосфорной кислотой) происходит поглощение паров воды фосфорной кислотой, а при значении РН2О < Р*Н2О - десорбuия.
Максимально допустимое общее давление (Рmax ) в системе может быть рассчитано следующим образом (при значении РН2О = Р*Н2О ; Р*Н2О над 84% -ной H3PO4 и 2800С равно 2,8МПа; отношение Н2О/С2Н4 = 0,7 и содержаниеинертных примесей в циркулирующем этилене - 15%): Рmax = РН2О + РС2Н4 + Рин. = 2,8 + (2.8/0,7)+(2,8/0,7*0,15)/0,85 = 7,5 МПа
Объёмная скорость. При циркуляционном процессе для его интенсификации и уменьшения количества побочных продуктов повышают объёмную скорость подачи сырья. При процессе прямой гидратации этилена это 1800-2000 час-1 , что соответствует времени контактирования 18-20 с. В данном случае, дальнейшее увеличение объёмной скорости приводит к возрастанию уноса фосфорной кислоты с носителя и уменьшению количества выделяющегося тепла, что не позволит проводить процесс в автотермическом режиме. Выход этанола за один проход в этих условиях составляет 5%, селективность по этилену 94-95 %
Свежий
и оборотный этилен сжимаются в компрессорах
от 1,2 до 8 МПа, смешиваются с водяным
паром, подогреваются в теплообменнике
4 теплом отходящей от реактора смеси и
перегреваются в трубчатой печи 3 до
2750С, после чего подаются в реактор-гидрататор
5. Перед входом в реактор в поток
вбрызгивается фосфорная кислота для
подпитки катализатора. Реактор
представляет собой полую колонну высотой
10 м и диаметром 1,5 м, работающую в режиме
идеального вытеснения. Для исключения
влияния коррозии от фосфорной кислоты
изнутри он выложен листами красной
меди. Реакционные газы содержат пары
унесенной фосфорной кислоты, которая
нейтрализуется гидроксидом натрия, а
образующиеся соли выделяются в
солеотделителе 6. Унос фосфорной кислоты
составляет 0,4 - 0,5 т/час с 1 м 3 катализатора.
Теплота отходящих реакционных газов
регенерируется в теплообменнике 4 для
нагрева входящей смеси. В холодильнике
7 происходит конденсация продуктов
реакции, а в сепараторе 8 разделяются
жидкие и газовые потоки. Вода, как менее
летучий компонент, конденсируется с
большей полнотой. Для дополнительного
выделения спирта из газа производится
его отмывка водой в абсорбере 9.
Непрореагировавший газ, содержащий
90-92% этилена, рециркулируют компрессором
2, а часть его сбрасывают, чтобы избежать
накопления примесей в системе. Отдувка
с оставляет
примерно 20% от введенного этилена и
направляется на установку газоразделения
для выделения этилена. Водный конденсат
после сепаратора 8 и жидкость из абсорбера
9 дросселируют (сбрасывают давление), в
результате чего выделяются растворенные
газы, отделяемые в сепараторе низкого
давления 10 и направляемые в топливную
линию. Жидкая фаза из сепаратора 10
представляет собой 15%ный водный раствор
этанола; содержащий примеси диэтилового
эфира, ацетальдегида и низкомолекулярных
полимеров этилена. Этот раствор подвергают
ректификации в ректификационных колоннах
11 и 12. В первой отгоняют наиболее летучий
диэтиловый эфир И ацетальдегид, а во
второй – этиловый спирт в виде азеотропной
смеси, содержащей 95% этанола и 5% воды.
Обогрев колонны осуществляется острым
паром. В кубе колонны 12 остается вода,
которую очищают от соли в ионообменной
установке 13 и возвращают на гидратацию.
При этом реализуется замкнутый цикл по
технологической воде, что позволяет
значительно снизить расход свежей воды,
исключить сброс отработанной воды в
стоки и сократить потери этилового
спирта. При необходимости получения
безводного этилового спирта ректификат
направляют в дегидрататор. В структуре
себестоимости спирта 30% приходится на
стоимость сырья.
оставляет
примерно 20% от введенного этилена и
направляется на установку газоразделения
для выделения этилена. Водный конденсат
после сепаратора 8 и жидкость из абсорбера
9 дросселируют (сбрасывают давление), в
результате чего выделяются растворенные
газы, отделяемые в сепараторе низкого
давления 10 и направляемые в топливную
линию. Жидкая фаза из сепаратора 10
представляет собой 15%ный водный раствор
этанола; содержащий примеси диэтилового
эфира, ацетальдегида и низкомолекулярных
полимеров этилена. Этот раствор подвергают
ректификации в ректификационных колоннах
11 и 12. В первой отгоняют наиболее летучий
диэтиловый эфир И ацетальдегид, а во
второй – этиловый спирт в виде азеотропной
смеси, содержащей 95% этанола и 5% воды.
Обогрев колонны осуществляется острым
паром. В кубе колонны 12 остается вода,
которую очищают от соли в ионообменной
установке 13 и возвращают на гидратацию.
При этом реализуется замкнутый цикл по
технологической воде, что позволяет
значительно снизить расход свежей воды,
исключить сброс отработанной воды в
стоки и сократить потери этилового
спирта. При необходимости получения
безводного этилового спирта ректификат
направляют в дегидрататор. В структуре
себестоимости спирта 30% приходится на
стоимость сырья.
5 .7.
Производство
ацетальдегида
Основные источники сырья для получения
ацетальдегида. Сравнение альтернативных
вариантов получения ацетальдегида.
Производство ацетальдегида окислением
этилена (Вакер-процесс) Стадии процесса.
Кинетика и механизм процесса.
Технологические схемы производства
ацетальдегида. Их сравнительный анализ.
Причины наличия циркуляции этилена в
однореакторной схеме производства
уксусного альдегида из этилена.
.7.
Производство
ацетальдегида
Основные источники сырья для получения
ацетальдегида. Сравнение альтернативных
вариантов получения ацетальдегида.
Производство ацетальдегида окислением
этилена (Вакер-процесс) Стадии процесса.
Кинетика и механизм процесса.
Технологические схемы производства
ацетальдегида. Их сравнительный анализ.
Причины наличия циркуляции этилена в
однореакторной схеме производства
уксусного альдегида из этилена.
Мировое производство ~ 1 млн.т /год
Ацетальдегид широко используется в химической промышленности в качестве полупродукта. Из него получают: уксусную кислоту, уксусный ангидрид, винилацетат, нбутиральдегид, этилацетат, пиридины, пентаэритрит, бутанол и 2-этилгексанол
Промышленные способы получения ацетальдегида
1) Гидратация ацетилена.
2) Дегидрирование и окислительное дегидрирование этилового спирта
3) Окисление этилена (наиболее эффективный способ)
П ромышленные
способы получения ацетальдегида
ромышленные
способы получения ацетальдегида
Гидратация ацетилена. Реакция открыта Кучеровым в 1881 году. Производство ацетальдегида этим способом началось в Германии в 1916 году
Классический синтез ацетальдегида по реакции Кучерова заключается во взаимодействии ацетилена с водой в присутствии окиси ртути, растворенной в серной кислоте. Процесс гидратации ацетилена проводится по непрерывной схеме: ацетилен барботируют через водный раствор катализатора (раствор сульфата ртути в серной кислоте). Содержание серной кислоты варьируется от 6 до 35%. Для предотвращения образования металлической ртути в контактный раствор добавляют окислитель – Fe2(SO4)3 . Процесс идёт при 90-95оС. Степень превращения ацетилена за один проход 30-50%. На выходе получают 8-10% раствор ацетальдегида. Недостатки этого метода – использование токсичного катализатора и дорогого сырья
Дегидрирование и окислительное дегидрирование этилового спирта на оксидных (ZnO, CuO, FeO) и металлических катализаторах (Ag, Cu, Au), соответственно.
С2Н5ОН → СН3СНО + Н2
С2Н5ОН + 0,5О2 → СН3СНО + Н2О
Д егидрирование
проводят при температуре 270-300оС,
селективность образования ацетальдегида
90-95%. Образующийся водород достаточно
чистый и может быть использован в
реакциях гидрирования. Дегидрирование
этанола в присутствии воздуха
(окислительное дегидрирование) включает
процесс сгорания водорода с выделением
тепла, необходимого для проведения
реакции дегидрирования. Пары этанола
смешивают с воздухом при 0,3 МПа и 450-550оС
и пропускают через серебряный катализатор.
Температура реакции контролируется
количеством подаваемого воздуха.
Селективность образования ацетальдегида
85-95%
егидрирование
проводят при температуре 270-300оС,
селективность образования ацетальдегида
90-95%. Образующийся водород достаточно
чистый и может быть использован в
реакциях гидрирования. Дегидрирование
этанола в присутствии воздуха
(окислительное дегидрирование) включает
процесс сгорания водорода с выделением
тепла, необходимого для проведения
реакции дегидрирования. Пары этанола
смешивают с воздухом при 0,3 МПа и 450-550оС
и пропускают через серебряный катализатор.
Температура реакции контролируется
количеством подаваемого воздуха.
Селективность образования ацетальдегида
85-95%
Окисление этилена (Вакер-процесс)
Более эффективный способ – окислением этилена был разработан в конце 50-х почти одновременно группой Смидта, Хафнера и др. (Консорциум электрохимической промышленности) и И.И. Моисеевым, М.Н. Варгафтиком и Я.К. Сыркиным (МИТХТ им. М.В. Ломоносова). Метод был основан на трех стехиометрических реакциях, которые в совокупности складывались в каталитический цикл. Cложение этих реакций приводит к итоговому уравнению каталитического процесса.
C2H4 + H2O + PdCl2 → CH3CHO + Pd(0) + 2HCl
Pd(0) + 2CuCl2 → PdCl2 + 2CuCl
2СuCl + 2HCl + 0,5O2 → 2CuCl2 + H2O
С2Н4 + 0,5O2 → CН3СНО
Это полифункциональная каталитическая система, в которой каждый из трех катализаторов (PdCl2 , CuCl2 , H2O) выполняет свою кинетическую функцию. Палладий (II) и нуклеофильный катализатор – вода – участвуют в стадиях окисления этилена в ацетальдегид. Медь (II) катализирует окисление восстановленного палладия кислородом. Процесс может проводиться с различными алкенами. Продуктами являются альдегиды и кетоны. Показатели зависят от природы используемого алкена. В случае этилена селективность – до 95%, побочные продукты – уксусная кислота – 2%, СО2 – 1%, хлорпроизводные – 1% (хлористый метил, хлористый этил, хлоруксусный альдегид). В случае окисления пропилена основным продуктом является ацетон. Селективность его образования достигает 90%. Пропионовый альдегид является побочным продуктом. Процесс внедрен в промышленное производство в 1960-е годы компаниями Waker-Chemie и Farbwerke Hoechst
Х арактеристика
процесса: сложный, необратимый,
экзотермический, гомогенно-каталитический
процесс
арактеристика
процесса: сложный, необратимый,
экзотермический, гомогенно-каталитический
процесс
Технологические схемы
Существует два технологических варианта проведения этого процесса – двухреакторный и однореакторный. В случае двухреакторной схемы в одном реакторе проводят первую и вторую реакции из трёх , а во втором реакторе - реакцию реокисления меди кислородом воздуха. В случае однореакторной схемы все три реакции протекают в одном аппарате.
Д вухреакторная
схема.
В Советском Союзе (России) была реализована
только двухреакторная схема. Процесс
в этом случае проводят при 100-110º и 1 МПа.
Этилен и водный раствор солей палладия
и меди подают в реактор типа «труба в
трубе» 3, который содержит инертную
насадку для увеличения межфазной
поверхности газ-жидкость. После реактора
3 контактный раствор вместе с реакционными
газами поступает в отпарную колонну 4.
Газовая фаза из колонны 4 (органические
продукты) попадает в ректификационную
колонну 5. Кубовый продукт колонны 5
используют для орошения колонны 4. Жидкую
фазу из колонны 4 (водный слой) перекачивают
в реактор 1, в котором происходит
реокисление солей меди воздухом.
Регенерированный каталитический раствор
через фазовый сепаратор 2 поступает в
реактор 3. Часть раствора из фазового
сепаратора 2 отводится в регенератор
10, в котором под воздействием хлористого
водорода при 150º происходит разложение
труднорастворимых оксалатов меди и
палладия (щавелевая кислота – один из
побочных продуктов). Это необходимо для
поддержания активности катализатора.
Газовая фаза из сепаратора 2 поступает
в скруббер 7, орошаемый, как и скруббер
6, кубовым продуктом из колонны 5. В
скруббере 6 промывается несконденсировавшийся
газ с верха колонны 5. Конденсат из
колонны 5, содержащий ацетальдегид,
через холодильник поступает на
ректификацию в колонну 8 для отделения
лёгких побочных продуктов (диоксид
углерода, хлористый метил, хлористый
этил), а затем в колонну 9 для отделения
более высококипящих продуктов
(хлорацетальдегид, у
вухреакторная
схема.
В Советском Союзе (России) была реализована
только двухреакторная схема. Процесс
в этом случае проводят при 100-110º и 1 МПа.
Этилен и водный раствор солей палладия
и меди подают в реактор типа «труба в
трубе» 3, который содержит инертную
насадку для увеличения межфазной
поверхности газ-жидкость. После реактора
3 контактный раствор вместе с реакционными
газами поступает в отпарную колонну 4.
Газовая фаза из колонны 4 (органические
продукты) попадает в ректификационную
колонну 5. Кубовый продукт колонны 5
используют для орошения колонны 4. Жидкую
фазу из колонны 4 (водный слой) перекачивают
в реактор 1, в котором происходит
реокисление солей меди воздухом.
Регенерированный каталитический раствор
через фазовый сепаратор 2 поступает в
реактор 3. Часть раствора из фазового
сепаратора 2 отводится в регенератор
10, в котором под воздействием хлористого
водорода при 150º происходит разложение
труднорастворимых оксалатов меди и
палладия (щавелевая кислота – один из
побочных продуктов). Это необходимо для
поддержания активности катализатора.
Газовая фаза из сепаратора 2 поступает
в скруббер 7, орошаемый, как и скруббер
6, кубовым продуктом из колонны 5. В
скруббере 6 промывается несконденсировавшийся
газ с верха колонны 5. Конденсат из
колонны 5, содержащий ацетальдегид,
через холодильник поступает на
ректификацию в колонну 8 для отделения
лёгких побочных продуктов (диоксид
углерода, хлористый метил, хлористый
этил), а затем в колонну 9 для отделения
более высококипящих продуктов
(хлорацетальдегид, у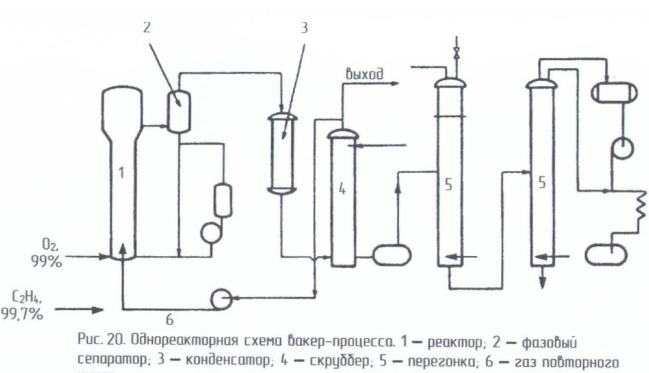 ксусная
и щавелевая кислоты). Тепло реакции (52
ккал/моль) используется для обогрева
колонны 4.
ксусная
и щавелевая кислоты). Тепло реакции (52
ккал/моль) используется для обогрева
колонны 4.
Однореакторная схема. Альтернативная схема использует один реактор, содержащий контактный раствор, в который подают высокочистые этилен и кислород. Процесс проводят при 120-130º и 0,3 МПа. Тепловой эффект снимают за счет испарения части воды. Убыль воды восполняется. Газообразные продукты, пройдя фазовый сеператор 2 и холодильник 3, поглощаются в скрубберах 4. Непрореагировавший этилен в избытке которого проводят процесс (выше верхнего предела взрываемости) рециркулируют обратно в реактор 1. Ацетальдегид очищают ректификацией в колоннах 5.
О сновной
проблемой двухреакторной схемы является
большое количество аппаратов, через
которые циркулирует коррозионноактивный
контактный раствор. Реакторы 1, 3, 10,
сепаратор 2, насосы, коммуникации
необходимо изготавливать из дорогих
коррозионностойких материалов (титановых
сплавов). Еще одним недостатком
двухреакторной схемы является низкая
производительность (~7 г/л·час). Преимущество
двухреакторной схемы над однореакторной
– безопасность, поскольку этилен и
кислород не контактируют между собой.
сновной
проблемой двухреакторной схемы является
большое количество аппаратов, через
которые циркулирует коррозионноактивный
контактный раствор. Реакторы 1, 3, 10,
сепаратор 2, насосы, коммуникации
необходимо изготавливать из дорогих
коррозионностойких материалов (титановых
сплавов). Еще одним недостатком
двухреакторной схемы является низкая
производительность (~7 г/л·час). Преимущество
двухреакторной схемы над однореакторной
– безопасность, поскольку этилен и
кислород не контактируют между собой.
Достоинства однореакторной схемы:
1) Мало аппаратов соприкасается с контактным раствором.
2) Производительность выше, чем в 2-х реакторном варианте.
Недостатки:
1) Необходимость использования чистого кислорода и этилена из-за рециркуляции газа.
2) Большой расход энергии на рециркуляцию газа.
3) Взрывоопасность.
4) Потери части этилена из-за отдувки газа из рецикла
Результаты сравнения показателей процессов получения ацетальдегида, проведенного во ВНИИОС, представлены в таблице (затраты на двухреакторный вариант приняты за 100%). Из этих данных следует, что однореакторная схема обладает существенными преимуществами перед двухреакторным вариантом и в отношении капитальных вложений, и по себестоимости ацетальдегида. Только потенциальная взрывоопасность ставит под сомнение целесообразность использования однореакторной схемы. Данные, приведенные в той же таблице и относящиеся к технологии получения ацетальдегида из ацетилена, свидетельствуют о неконкурентоспособности этого варианта в основном из-за высокой стоимости сырья и несоответствия экологическим требованиям.