

Патологическая анатомия Серов
.pdfБолезни прогенеза и киматогенеза
Гаметопатии
Гаметопатии - это патология гамет. К ним относятся любые повреждения яйцеклетки и сперматозоида во время ово- и сперматогенеза до оплодотворения. Понятие «гаметопатии» охватывает все виды повреждения мужской и женской гаметы: мутации генов и возникновение наследственных болезней и наследственных пороков развития, хромосомные аберрации с возникновением чаще не наследуемых хромосомных болезней, геномные мутации - изменения числа хромосом гаметы, обычно приводящие к самопроизвольному аборту или хромосомной болезни. Кроме того, необходимо учитывать, что тяжелые повреждения не только ядра, но и цитоплазмы гаметы становятся источником их гибели с развитием стерильности и бесплодия или спонтанных абортов и выкидышей. Из этого следует, что гаметопатии являются одним из факторов внутриутробной летальности, не поддающейся пока точной регистрации.
При повреждении ядра гаметы могут происходить изменения генетического аппарата. Изменения генов, их мутации приводят к закреплению этих изменений в последующих клеточных генерациях. Следует учитывать, что гаметы являются носителями генов, унаследованных ими от всех отдаленных предков. Поэтому в понятие гаметопатии входит поражение не только гамет родителей, но и более отдаленных предков пробанда. Гамета с дефектом гена или генов может стать источником наследственных пороков развития или заболеваний, проявляющихся на разных этапах внутриутробного и внеутробного развития.
Генные пороки и болезни могут наследоваться по аутосомно-рецессивному, аутосомно-
доминантному типамили мутантный ген может быть сцеплен с половой Х-хромосомой. При аутосомно-рецессивном типе наследования у пробанда воз-
никает порок только в том случае, если мутантный ген был получен и от отца, и от матери. Родители пробанда сами могут быть здоровы, являясь лишь гетерозиготными носителями мутантного гена. При аутосомно-доминантном типе наследования мутантный ген передается от отца или от матери, которые сами страдают аналогичным пороком.
Пороки, гены которых локализованы в Х-хромосоме, в свою очередь могут наследоваться по рецессивному или доминантному типу. Пороки, сцепленные с Х-хромосомой, передающиеся по рецессивному типу, наблюдаются, как правило, у мальчиков, так как единственная у них Х- хромосома является пораженной. Мутантный ген передает мать, не являющаяся больной. Очень редко носительницей порока может быть девочка. Это бывает в том случае, если отец являлся больным, а мать - носительницей мутантного гена.
Кроме локального поражения генетического аппарата ядра гаметы вследствие мутации генов, в период гаметогенеза может появляться мутация хромосом в виде изменений их числа и структуры. Мутации хромосом получили название хромосомных аберраций. Хромосомные аберрации возникают чаще всего в момент редукционного деления гамет. Их следствием
711
являются хромосомные болезни, которые, однако, в большинстве случаев не наследуются, так как их носители чаще умирают в детстве или являются бесплодными.
Типичными примерами хромосомных болезней являются болезнь Дауна (трисомия по 21-й паре аутосом),синдром Патау (трисомия по 13- 15-й паре аутосом), синдром ШерешевскогоТернера (моносомия половой хромосомы - 45 ХО) и др.
Болезнь Дауна, наблюдающаяся у новорожденных в соотношении 1:600, 1:700, встречается наиболее часто. Клинически у детей с рождения отмечается выраженная задержка умственного и физического развития. Больные имеют типичный внешний вид: косой разрез глаз, западающая спинка носа, высокое небо, низкое расположение маленьких ушных раковин, выраженная гипотония мышц. Дети умирают чаще от интеркуррентных заболеваний. У большинства из них обнаруживаются пороки развития сердца и магистральных сосудов (тетрада Фалло и др.), реже - пороки развития пищеварительной и мочеполовой систем. У этих детей отмечаются недоразвитие полушарий большого мозга, особенно лобных его долей с задержкой дифференцировки нейронов, нарушения процессов миелинизации, архитектоники кровеносных сосудов мозга.
Синдром Патау у новорожденных и мертворожденных встречается с частотой 1 на 5149 рождений. Характерны выраженная общая гипоплазия, аномалии черепа и лица: низкий скошенный лоб, узкие глазные щели, запавшее переносье, широкое основание носа, гипотелоризм, «дефекты скальпа», низко расположенные деформированные ушные раковины, типичные расщелины верхней губы и неба. Отмечаются полидактилия и флексорное положение кистей, микрофтальмия, колобома и помутнение роговицы. Со стороны головного мозга отмечаются микроцефалия, аринэнцефалия (отсутствие обонятельного мозга), аплазия или гипоплазия червя мозжечка и др. Отмечаются также врожденные пороки сердца, органов пищеварения, мочевой системы и др. Дети нежизнеспособны.
Бластопатии
Бластопатия - патология бластоцисты, возникающая в период нидации и дробления в первые 15 дней от момента оплодотворения до выделения эмбрио-и трофобласта.
Этиология и патогенез. Причиной бластопатии чаще всего являются хромосомные аберрации в сочетании с влияниями среды (эндокринные заболевания матери, гипоксия и др.). Патогенез зависит от вида поражения бластоцисты. Так, например, патогенез двойниковых уродств связан с появлением во время дробления двух или более самостоятельно растущих центров. Полагают, что если эти центры разобщены друг с другом, то развиваются два независимо растущих однояйцевых близнеца, нормальное развитие которых не следует относить к бластопатиям. Если центры роста расположены близко и имеют общую для двух близнецов промежуточную зону, то развиваются два сросшихся близнеца. В обоих случаях возможно развитиесимметричных и асимметричных близнецов.
712
Морфология бластопатии разнообразна. К ним относятся нарушения имплантации бластоцисты, а именно эктопическая беременность, поверхностная или очень глубокая имплантация бластоцисты в эндометрий, нарушение ориентации формирующегося эмбриобласта в бластоцисте по отношению к эндометрию, аплазия или гибель развивающегося эмбриобласта с образованием пустого зародышевого мешка, пороки развития всего эмбриона, некоторые одиночные пороки, двойниковые уродства и, наконец, аплазия или гипоплазия формирующегося трофобласта - амниона, амниотической ножки,
желточного мешка. Поверхностная или чрезмерно глубокая имплантация бластоцисты приводит к порокам развития формы, локализации, а также приращению плаценты (см. ниже), которые чреваты гибелью плода во время акта родов. Нарушения
ориентации эмбриобласта при полной топографической инверсии заканчиваются гибелью эмбриобласта. При неполной инверсии наблюдаются пороки развития пуповины (см. ниже), которые могут приводить к гибели плода во время родов. Пустые зародышевые мешки представляют собой бластоцисты, не содержащие эмбриобласт или содержащие его
остатки. Иногда в них можно обнаружить амниотические оболочки, пуповину, желточный мешок.
Патология развития всего эмбриона представляет собой общие тяжелые нарушения, не совместимые с жизнью.
Одиночные и множественные пороки развития, возникающие в период бластулы (в первые 8- 12 нед), встречаются в 14,3-22,9% всех спонтанно абортированных зародышей. При этом в 46,2% случаев они сопровождаются аномалиями последа. Такое сочетание часто приводит к гибели зародыша.
Двойниковые уродства встречаются в виде сросшейся двойни. Если сросшаяся двойня состоит из равных симметрично развитых компонентов, она называется диплопагусом (diplopagus от греч. diplos - двойной, agus - соединять); если же она состоит из асимметрично развитых ком-
понентов - гетеропагусом (heteropagus от греч. heteros - другой), при этом недоразвитый близнец, находящийся в зависимости от другого, развитого, получил название паразита. Для обозначения локализации сращения близнецов к анатомическому названию места сращения добавляют также слово пагус; например, сращение в области головы называют краниопагусом, в области груди - торакопагусом, в области таза -ишиопагусом и др.
Двойниковые уродства сочетаются с нежизнеспособностью. В редких случаях описана значительная продолжительность жизни таких близнецов до зрелого возраста. В легких случаях сращений только мягких тканей возможна хирургическая коррекция.
Эмбриопатии
713
Эмбриопатия - патология эмбрионального периода с 16-го дня беременности до 75-го дня включительно, в течение которого заканчивается основной органогенез и формирование амниона и хориона. К основным видам эмбриопатий относят врожденные пороки развития.
Врожденным пороком развития называют стойкое морфологическое изменение органа, части тела или всего организма, выходящее за пределы вариаций нормального строения определенного биологического вида, возникащее внутриутробно в результате нарушений морфогенеза. Так как органогенез завершается в основном в эмбриональный период, большинство пороков развития появляется именно на этом этапе внутриутробного существования. Однако, кроме врожденных пороков с нарушениями основного морфогенеза органов или частей тела, имеются врожденные пороки, при которых нарушения развития наблюдаются на уровне тканевой дифференцировки. Они часто бывают системными, например пороки развития поперечнополосатой мускулатуры (врожденная миатония Оппенгейма), соединительной ткани (болезнь Марфана), кожи (врожденный ихтиоз), костей хрящевого генеза (врожденная хондродисплазия) и др. Пороки развития могут касаться также тканей одного органа, например гипоплазия гладкой мышечной ткани
при megaureter, нервного интрамурального аппарата - при megacolon, легочной ткани - при кистозном легком и др. По срокам возникновения эти пороки относятся к ранним фетопатиям. Ранние фетопатии часто сочетаются с эмбриопатиями; например, врожденный ихтиоз и хондродисплазия - с пороками развития лица, болезнь Марфана - с пороками развития лица и аорты и др. Частота врожденных пороков, по данным ВОЗ, составляет 1,3% от общего числа рождений.
Любой врожденный порок может проявляться в виде: 1) отсутствия какого-либо органа или части тела (агенезия, аплазия); 2) недоразвития органа (гипоплазия); 3) чрезмерного развития (гиперплазия) или наличия избыточного числа органов (удвоение и др.); 4) изменения формы (слияние органов, атрезия, стеноз отверстий, каналов, дизрафия - незаращение эмбриональных щелей, экстрофия - выворот и др.); 5) изменения
в расположении органов (эктопия); 6) персистирования эмбриональных провизорных (предсуществовавших) органов.
Классификация. Врожденные пороки развития разделяют по степени распространенности в организме, по локализации в том или ином органе, по этиологии. По распространенности врожденные пороки могут быть: 1) изолированными - с поражением одного органа; 2) системными - с поражением нескольких органов одной из систем; 3)
множественными - с поражением органов разных систем. По локализации различают пороки развития центральной нервной, сердечно-сосудистой, пищеварительной, мочеполовой и других систем. Врожденные пороки развития названной локализации имеют наибольшее значение в патологии. Чаще всего встречаются пороки развития центральной нервной и сердечно-сосудистой систем, так как именно эти системы имеют наибольший тератогенный терминационный период (см. рис. 292). Изолированные пороки развития встречаются чаще множественных, несмотря на то что тератогенный терминационный период для многих органов во времени совпадает.
714
Наиболее совершенной является классификация пороков развития по этиологии, однако уровень современных знаний пока не позволяет ее придерживаться. Однако известны отдельные виды системных и множественных врожденных пороков, связанных с определенной этиологией, например рубеолярная эмбриопатия, алкогольная, талидомидная эмбриопатии и др., а также наследственно обусловленные генотипические врожденные пороки и врожденные пороки вследствие хромосомных аберраций; последние, как правило, носят характер множественных.
Разграничение генотипических врожденных пороков с их фенокопиями возможно с помощью генеалогического метода изучения родословной, цитогенетического метода, позволяющего изучить кариотип тканей носителя порока при их культивировании, с помощью близнецового метода, основанного на частоте выявления врожденных пороков у однояйцевых близнецов и метода дерматоглифики - изучения комплекса кожных узоров, расположенных на ладонях, подошвах и сгибательной поверхности пальцев, который используется для срочной диагностики хромосомных болезней.
Врожденные пороки центральной нервной системы
Врожденные пороки ЦНС по частоте занимают первое место среди других пороков, встречаются в 30% случаев среди пороков развития, обнаруживаемых у детей.
Этиология и патогенез. Из экзогенных факторов точно установлено значение вируса краснухи, иммунодефицита человека, простого герпеса, предполагается влияние вирусов цитомегалии, Коксаки, лекарственных препаратов (хинин, гидантоин и др.), алкоголя, лучевой энергии, гипоксии. Несомненное значение имеют генные мутации; при хромосомных болезнях в числе множественных пороков они встречаются почти как правило. Развитие порока связано с воздействием повреждающего агента в течение всего эмбрионального периода, включая ранний фетальный.
Наиболее тяжелые пороки возникают при повреждении в начале закладки нервной трубки (3- 4-я неделя внутриутробной жизни).
Патологическая анатомия. К основным наиболее тяжелым видам врожденных пороков ЦНС относятся следующие. Анэнцефалия - агенезия головного мозга, при которой отсутствуют передние, средние, иногда и задние его отделы. Продолговатый и спинной мозг сохранены. На месте головного мозга обнаруживается соединительная ткань, богатая сосудами, в которой встречаются отдельные нейроны и клетки нейроглии. Анэнцефалия сочетается с акранией - отсутствием костей свода черепа, покрывающих их мягких тканей и кожи.
Микроцефалия - гипоплазия головного мозга, уменьшение его массы и объема; сочетается с одновременным уменьшением объема черепной коробки и утолщением костей черепа; возможны разные степени тяжести этого порока. Микрогирия - увеличение числа мозговых извилин наряду с уменьшением их величины.
715

Порэнцефалия - появление кист различных размеров в головном мозге, сообщающихся с боковыми желудочками мозга, выстланных эпендимой. От истинной порэнцефалии следует отличать ложную, при которой кисты не сообщаются с путями оттока ликвора и образуются на месте бывших размягчений ткани головного мозга.
Врожденная гидроцефалия - избыточное накопление ликвора в желудочках мозга (внутренняя гидроцефалия)или в субарахноидальных пространствах (наружная
гидроцефалия) (рис. 294) сопровождается увеличением мозгового черепа и резким несоответствием его с лицевым - лицо кажется маленьким, лоб - нависшим. Наблюдаются расхождение и ис-
Рис.
294. Гидроцефалия (по А.В. Цинзерлингу)
тончение костей черепа, выбухание родничков. Нарастает атрофия вещества головного мозга, в большинстве случаев связанная с нарушениями оттока ликвора вследствие стеноза, раздвоения или атрезии водопровода большого мозга (сильвиева водопровода), атрезии срединных и боковых отверстий IV желудочка и межжелудочкового отверстия.
Циклопия - редкий порок, характеризующийся наличием одного или двух глазных яблок, расположенных в одной глазнице, с одновременным пороком развития носа и обонятельной доли головного мозга. Назван изза сходства лица плода с лицом мифического чудовища - циклопа.
Грыжи головного и спинного мозга представляют собой выпячивание вещества мозга и его оболочек через дефекты костей черепа, их швов и позвоночного канала. Грыжи головного мозга: при наличии в грыжевом мешке только оболочек головного мозга и ликвора носят
716
название менингоцеле, оболочек и вещества мозга -менингоэнцефалоцеле, вещества мозга и мозговых желудочков - энцефалоцистоцеле. Чаще встречаются грыжи спинного мозга, связанные с расщеплением дорсальных отделов позвонков, которые называются spina
bifida. Грыжи спинного мозга, как и головного, в зависимости от содержимого грыжевого мешка можно разделять на менингоцеле, миелоцеле, менингомиелоцеле. Очень редко встречается рахиосхиз - полный дефект задней стенки позвоночного канала, мягких тканей, кожи и мозговых оболочек; при этом распластанный спинной мозг лежит открытым на передней стенке канала, выпячивания нет.
Прогноз при врожденных пороках ЦНС неблагоприятен, большинство из них несовместимы с жизнью. Хирургическая коррекция эффективна только в некоторых случаях мозговых и спинномозговых грыж. Дети умирают часто от присоединения интеркуррентных инфекционных заболеваний. Мозговые и спинномозговые грыжи осложняются гнойной инфекцией с развитием гнойного менингита и менингоэнцефалита.
Врожденные пороки сердца
Врожденные пороки сердца по частоте занимают второе место после пороков развития ЦНС. По данным разных авторов, они встречаются в 16-40% среди других пороков и в 3-8% случаев по данным вскрытий детей, умерших в перинатальном периоде.
Этиология и патогенез. Причины этих пороков разнообразны и не связаны с влиянием какихлибо определенных экзогенных факторов. Имеют несомненное значение генные мутации и хромосомные аберрации. Среди множественных пороков, наблюдающихся при хромосомных болезнях, пороки сердца встречаются реже, чем пороки ЦНС. Развитие порока связано с воздействием повреждающего агента на эмбрион от 3-й до 11-й недели внутриутробного развития. Различные виды пороков зависят от искажения этапов морфогенеза сердца, из которых основными являются дефекты первоначально парных закладок сердца, неправильные изгибы первичной сердечной трубки, задержка развития или неправильное расположение перегородок сердца, делящих его и артериальный ствол на
правую и левую половины, персистирование предсердно-желудочковых соединений, существующих во время внутриутробной жизни.
Патологическая анатомия. При врожденных пороках сердца в процессе гипертрофии миокарда у детей в возрасте первых 3 мес жизни участвуют не только увеличение объема мышечных волокон с гиперплазией их ультраструктур, но и истинная гиперплазия кардиомиоцитов. Одновременно с этим развивается гиперплазия ретикулиновых аргирофильных волокон стромы сердца. Последующие дистрофические изменения миокарда и стромы, вплоть до развития микронекрозов, приводят к постепенному разрастанию соединительной ткани и возникновению диффузного и очагового кардиосклероза.
Компенсаторная перестройка сосудистого русла гипертрофированного сердца сопровождается увеличением в нем интрамуральных сосудов, артерио-венозных
717
анастомозов, наименьших вен (так называемых сосудов Вьессена-Тебезия) сердца. В связи со склеротическими изменениями в миокарде, а также усилением кровотока в его полостях появляется утолщение эндокарда за счет разрастания в нем эластических и коллагеновых волокон. Перестройка сосудистого русла развивается также и в легких. У детей с врожденными пороками сердца наблюдается отсталость общего физического развития.
Смерть наступает в первые дни жизни от гипоксии при особо тяжелых формах пороков или позже от развития сердечной недостаточности. С прогрессом грудной хирургии стало возможным лечение многих врожденных пороков с использованием хирургической коррекции и протезирования, что заметно изменило течение и исходы врожденных пороков сердца у детей. Благодаря сложности процессов эмбриогенеза сердца врожденные пороки его разнообразны. Однако большинство из них связано с ненормальными сообщениями между малым и большим кругом кровообращения, сужениями в этих системах или с отсутствием нормальных сообщений между ними, вплоть до несовместимого с жизнью полного разобщения малого и большого круга кровообращения. В зависимости от степени гипоксии, обусловленной уменьшением кровотока в малом круге кровообращения и направлением тока крови через ненормальные пути между малым и большим кругом кровообращения, пороки сердца могут быть разделены на два основных типа - синий и белый. При пороках синего типа отмечаются уменьшение кровотока в малом круге кровообращения, гипоксия и направление тока крови по анормальному пути - справа налево. При пороках белого типа гипоксия отсутствует, направление тока крови слева направо. Однако это деление схематично и не всегда применимо ко всем типам врожденных пороков сердца.
Врожденные пороки с нарушением деления полостей сердца. Дефект межжелудочковой перегородкивстречается часто, возникновение его зависит от отставания в росте одной из структур, формирующих перегородку, вследствие чего между желудочками развивается ненормальное сообщение. Чаще наблюдается дефект в верхней соединительнотканной (мембранозной) части перегородки (рис. 295). Кровоток через дефект
718
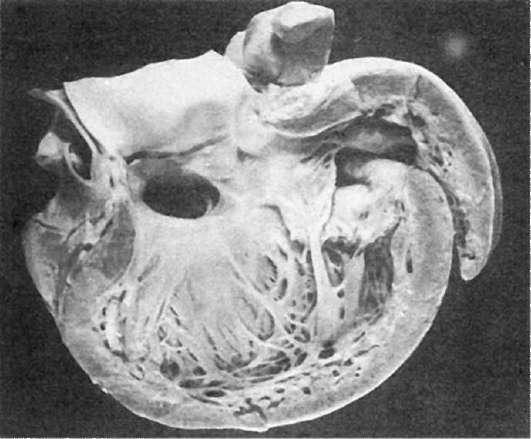
Рис.
295. Дефект в межжелудочковой перегородке сердца (по А.В. Цинзерлингу)
осуществляется слева направо, поэтому цианоза и гипоксии не наблюдается (белый тип порока). Степень дефекта может варьировать, вплоть до полного отсутствия перегородки. При значительном дефекте развивается гипертрофия правого желудочка сердца, при незначительном - существенных изменений гемодинамики не происходит.
Дефект межпредсердной перегородки в виде изолированного порока встречается редко. Он возникает либо при нарушениях развития первичной предсердной перегородки на 5-й неделе эмбриогенеза, либо позднее, при формировании вторичной перегородки и овального отверстия. Дефект первичной перегородки имеет вид отверстия, расположенного непосредственно над клапанами желудочков; при дефекте вторичной перегородкиимеется широко открытое овальное отверстие, лишенное заслонки. В том и в другом случае ток крови происходит слева направо, гипоксии и цианоза не бывает (белый тип порока). Переполнение кровью правой половины сердца сопровождается гипертрофией правого желудочка и расширением ствола и ветвей легочной артерии. Полное отсутствие межжелудочковой или межпредсердной перегородок приводит к развитию трехкамерного сердца - тяжелого порока, при котором, однако, в период компенсации не наблюдается полного смешения артериальной и венозной крови, так как основной ток той или другой крови сохраняет свое направление и поэтому степень гипоксии нарастает по мере прогрессирования декомпенсации.
Врожденные пороки сердца с нарушениями деления артериального ствола. Общий артериальный ствол при полном отсутствии деления артериального ствола встречается редко. При этом пороке один общий артериальный
719
ствол берет свое начало от обоих желудочков, у выхода располагается 4 полулунных клапана или меньше; порок часто сочетается с дефектом межжелудочковой перегородки. Легочные артерии отходят от общего ствола недалеко от клапанов, до ответвления крупных сосудов головы и шеи, они могут совсем отсутствовать и тогда легкие получают кровь из расширенных бронхиальных артерий. При этом пороке наблюдаются резкая гипоксия и цианоз (синий тип порока), дети нежизнеспособны.
Полная транспозиция легочной артерии и аорты возникает при неправильном направлении роста перегородки артериального ствола, когда она растет не по спирали, а в направлении, противоположном остальным, нормально развивающимся отделам сердца. При этом пороке аорта помещается спереди и справа от правого желудочка сердца, легочная артерия лежит позади аорты и отходит от левого желудочка. Артериальная кровь может попасть в большой круг кровообращения только при дефектах в перегородках сердца или при незаращении артериального (боталлова) протока и овального отверстия. Порок сопровождается резкой гипоксией и цианозом (синий тип порока).
Значительно страдает миокард, так как венечные артерии не получают артериальной крови. Дети нежизнеспособны.
Стеноз и атрезия легочной артерии наблюдаются при смещении перегородки артериального ствола вправо, часто сочетаются с дефектом межжелудочковой перегородки и другими пороками. При значительном сужении легочной артерии кровь в легкие попадает через артериальный (боталлов) проток и расширяющиеся бронхиальные артерии. Порок сопровождается гипоксией и выраженным цианозом (синий тип порока).
Стеноз и атрезия аорты являются следствием смещения перегородки артериального ствола влево. Они встречаются реже, чем смещение перегородки вправо, часто сопровождаются гипоплазией левого желудочка сердца. При этом наблюдаются резкая степень гипертрофии правого желудочка сердца, расширение правого предсердия и резкий общий цианоз. Дети нежизнеспособны.
Сужение перешейка аорты (коарктация), вплоть до его атрезии, компенсируется развитием коллатерального кровообращения через межреберные артерии, артерии грудной клетки и резкой гипертрофией левого желудочка сердца.
Незаращение артериального (боталлова) протока можно считать пороком при наличии его с одновременным расширением у детей старше 3 мес жизни. Ток крови осуществляется при этом слева направо (белый тип порока). Изолированный порок хорошо поддается хирургической коррекции.
Комбинированные врожденные пороки сердца. Среди комбинированных пороков чаще встречаются триада, тетрада и пентада Фалло. Триада Фалло имеет 3 признака: дефект межжелудочковой перегородки, стеноз легочной артерии и как следствие этого гипертрофия правого желудочка. Тетрада Фалло имеет 4 признака: дефект межжелудочковой перегородки,
720