

Травень В.Ф. - Органическая химия. В 3 т. Т. 1
..pdf1.13. Концепция механизма органической реакции |
101 |
Рис. 1.9. Энергетическая диаграмма реакции гидробромирования этилена: 1 — π-ком- плекс, 2 — ионная пара
ции G≠ реакций. Величина G ° определяет лишь вероятность протекания как обратимых, так и необратимых реакций. Скорость реакции зависит только от свободной энергии активации G≠ и не зависит от G °. Значения G≠ для большинства органических реакций находятся в интервале 418–
1254 кДж/моль (10–30 ккал/моль).
Только немногие органические реакции протекают в одну стадию. Большинство органических реакций многостадийны и имеют несколько переходных состояний. Ряд примеров были показаны выше среди кислотно-ос- новных реакций Льюиса.
Рассмотрим подробнее реакцию гидробромирования этилена. Эта реакция протекает с образованием двух промежуточных соединений — π-комплекса и карбокатиона, находящегося, вероятнее всего, в составе ионной пары.
|
|
|
|
|
|
|
HBr |
|
|
|
|
|
|
|
|
|||
CH2 |
|
CH2 |
+ HBr |
|
|
|
CH2 |
|
|
CH2 |
|
|
|
CH3 |
|
CH2Br |
|
CH3CH2Br |
|
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
||||||||
этилен |
бромо- |
|
π-комплекс |
|
|
|
ионная пара |
|
бромэтан |
|||||||||
|
|
|
водород |
|
|
|
|
|
|
|
|
|
|
|
|
|
||
Стадии образования π-комплекса соответствует переходное состояние ПС1 (рис. 1.9). Эта стадия, как правило, вносит незначительный вклад в энергетику процесса и иногда на энергетической диаграмме ее не обозначают.
Переходное состояние ПС2 соответствует образованию этил-катиона в составе ионной пары, а переходное состояние ПС3 — дальнейшему превращению этил-катиона — присоединению бромид-аниона. Этил-катион является, таким образом, промежуточным соединением реакции гидробромирования этилена; его энергии соответствует впадина между состояниями ПС2 и ПС3 на энергетической кривой.

102 Глава 1. Природа ковалентной связи. Электронные эффекты. Кислоты и основания
Промежуточное соединение, в отличие от активированного комплекса (переходное состояние), даже будучи нестабильным, может быть в определенных условиях изучено экспериментально, в том числе спектральными методами.
Более высокий энергетический барьер отвечает более медленной стадии. Самый высокий барьер соответствует наиболее медленной стадии, которая называется скоростьлимитирующей стадией. В случае двухстадийного процесса энергетическая диаграмма может иметь различный вид в зависимости от того, первая или вторая стадия требует большей энергии активации и, следовательно, лимитирует скорость всего превращения.
 ОБРАТИТЕ ВНИМАНИЕ!
ОБРАТИТЕ ВНИМАНИЕ!
Механизмом реакции называют подробное (стадия за стадией) описание пути, по которому реагенты превращаются в продукты. Это описание включает перемещение электронов, которое ведет к образованию и разрыву связей, а также пространственные отношения между реагентами в ходе реакции.
Переходное состояние (активированный комплекс) — максимум на энер-
гетическом профиле реакции, группировка атомов, находящаяся в переходном состоянии, а промежуточное соединение (интермедиат) — частица, энергия
которой соответствует минимуму энергии, расположенному на энергетической кривой между двумя максимумами; промежуточными соединениями часто выступают карбокатионы, карбанионы, радикалы.
1.13.2.Кинетика
Важнейший инструмент изучения механизма — анализ кинетики органической реакции, поскольку она определяет число и природу молекул, включаемых в скоростьлимитирующую стадию.
Измерение скорости проводят при фиксированной температуре и контролируемых концентрациях реагентов. Измеряя снижение концентраций исходных реагентов и рост концентраций продуктов в зависимости от времени, получают кинетическое уравнение. Кинетическое уравнение связывает скорость реакции с концентрациями исходных реагентов A и B через константу скорости (k):
w = k [A][B].
Зависимость скорости от концентрации реагента определяет порядок реакции по данному реагенту. В реакции первого порядка скорость зависит от концентрации только одного реагента в первой степени. В реакции второго порядка скорость зависит от квадрата концентрации одного реагента или

1.13. Концепция механизма органической реакции |
103 |
пропорциональна концентрациям двух реагентов в первой степени. Например, реакция гидролиза хлорметана под действием гидроксид-иона [см. уравнение (4) в разд. 1.9.1] имеет второй порядок.
w = — |
d[CH3Cl] |
= |
d[CH3OH] |
= k |
[CH Cl][ OH]. |
|
|||||
|
dτ |
|
dτ |
2 |
3 |
|
|
|
|
Термин «порядок реакции» следует отличать от термина «молекулярность реакции». Молекулярность реакции зависит от числа частиц, участвующих в образовании активированного комплекса. Реакция с молекулярностью 1 называется мономолекулярной, с молекулярностью 2 — бимолекулярной и 3 — тримолекулярной. Молекулярность реакции, в отличие от порядка реакции, определяется не из кинетических данных, а нашим представлением о механизме реакции. В то же время порядок и молекулярность реакции могут и совпадать.
Согласно теории переходного состояния, переходное состояние реакции можно рассматривать как псевдомолекулярную частицу, обладающую всеми свойствами, присущими молекулам вещества, и находящуюся в равновесии с исходными реагентами. Аналогично любой другой константе равновесия константа равновесия K≠ должна удовлетворять основному уравнению равновесного состояния
G≠ = –RT lnK≠, или K≠ = e– G≠/RT,
где R — газовая постоянная, T — абсолютная температура.
Эти уравнения ставят константу равновесия K≠ в зависимость от свободной энергии активации G≠. На основе квантово-статистических методов между константой скорости реакции (k) и константой равновесия (K≠) выведено соотношение
kБT ≠ k = K ,
h
где kБ — константа Больцмана (R/NА), h — постоянная Планка, NА — число Авогадро.
Величина k |
Б |
T/h является универсальной; ее значение равно 6 1012 |
|
(при 300 K) для любой реакции. Подставляя выражение для K≠ в получен- |
|||
ное уравнение, имеем: |
|||
kБT |
– |
G≠/(RT ) |
|
k = e |
|
|
. |
h |
|
|
|
Согласно этому выражению, константа скорости реакции является функцией работы (– G≠), необходимой для перевода молекул из исходного состояния в переходное состояние.

104 Глава 1. Природа ковалентной связи. Электронные эффекты. Кислоты и основания
Продолжая применять принципы термодинамики к переходному состоянию, можно записать
G≠ = H≠ – T S≠.
Свободная энергия активации G≠ образована, таким образом, энтальпией активации H≠ и энтропией активации S≠.
Энтропия вещества, как и энтальпия, является одним из его характерных свойств. Физическая суть энтропии — мера неупорядоченности системы. Системы с высокой неупорядоченностью (низкоорганизованные системы) характеризуются большими значениями энтропии; низкая энтропия характерна для высокоорганизованных систем. Поэтому, если процесс сопровождается положительным изменением энтропии ( S > 0), член T S дает отрицательный вклад в G≠. В этом случае переходное состояние менее упорядоченно, чем исходные вещества, процесс будет стремиться проходить спонтанно.
Часто вклад энтропийного члена невелик, и свободную энергию актива-
ции G≠ оценивают величиной H≠ или энергии активации E : |
||
|
|
акт |
G≠ ~ H≠ = E |
акт |
– RT. |
- |
|
|
Энергия активации, в свою очередь, легко может быть найдена из температурной зависимости константы скорости реакции
dlnk/dT = Eакт/RT2
или, в интегрированной форме,
k = Ae–Eакт/(RT ).
Измеряя скорость реакции при нескольких температурах и нанося на гра-
фик значения ln k (по оси ординат) и 1/T (по оси абсцисс), величину Еакт/R получают в виде тангенса угла наклона прямой в указанных координатах.
1.13.3.Кинетический контроль органической реакции
Понимание факторов, определяющих скорости органических реакций, позволяет ввести понятие кинетический контроль органической реакции. Чтобы лучше понять, в чем суть кинетического контроля, рассмотрим реакцию гидробромирования пропена — ближайшего аналога этилена
 CH3 CH2 CH2 Br
CH3 CH2 CH2 Br
1-бромпропан
CH3 CH CH2 HBr
CH2 HBr
Br
CH3 CH CH3
2-бромпропан

1.13. Концепция механизма органической реакции |
105 |
Рис. 1.10. Энергетическая диаграмма реакции гидробромирования пропена
и попытаемся ответить на вопрос, почему продуктом этой реакции является 2-бромпропан, а не 1-бромпропан?
Энергетическая диаграмма гидробромирования пропена показана на рис. 1.10. По-видимому, и эта реакция начинается с образования π-ком- плекса:
HBr
CH3 CH |
|
|
CH2 + HBr |
|
CH3 CH |
|
|
|
CH2 |
|
|
|
|
|
|
||||
|
|
|
|
|
|||||
|
|
|
|
|
|
||||
пропен |
бромоводород |
π-комплекс |
|||||||
Однако переход π-комплекса в σ-комплекс в этом случае может идти по двум направлениям:
а — с образованием изопропил-катиона (Па) в качестве промежуточного соединения (протон присоединяется к С1-атому) и 2-бромпропана как продукта реакции;
б — с образованием н-пропил-катиона (Пб) в качестве промежуточного соединения (протон присоединяется к С2-атому) и 1-бромпропана как продукта реакции.
|
a |
|
|
|
|
|
|
|
|
Br |
||||
HBr |
CH3 |
|
CH |
|
CH3 + Br |
|
CH3 |
|
|
|
CH |
|
CH3 |
|
|
|
|
|
|
||||||||||
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
Па |
|
1-бромпропан |
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|||
CH3 CH
CH CH2
CH2
б |
CH |
|
CH |
|
CH |
+ Br |
|
CH |
|
CH |
|
CH |
|
Br |
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
||||||||
3 |
2 |
2 |
|
3 |
2 |
2 |
|
|
||||||
Пб |
2-бромпропан |
|

106 Глава 1. Природа ковалентной связи. Электронные эффекты. Кислоты и основания
Как уже отмечалось в разд. 1.12.3, устойчивость изопропил-катиона по сравнению с н-пропил-катионом значительно выше:
CH3 |
|
CH |
|
CH3 |
CH3 |
|
CH2 |
|
CH2 |
|
|
|
|
||||||
две электронодонорные группы |
одна электронодонорная группа |
||||||||
у катионного центра |
у катионного центра |
||||||||
Из диаграмм, представленных на рис. 1.9 и 1.10, следует, что именно устойчивость промежуточного карбокатиона определяет высоту активационного барьера, т. е. свободную энергию активации реакции гидробромирования алкена. Вследствие большей устойчивости вторичного карбокатиона 2-бромпропан и оказывается практически единственным продуктом взаимодействия пропена и HBr. Речь идет в этом случае о кинетическом контроле реакции.
 ОБРАТИТЕ ВНИМАНИЕ!
ОБРАТИТЕ ВНИМАНИЕ!
Кинетически-контролируемой реакцией называют реакцию, состав продуктов в которой определяется активационными параметрами ( G8, H8, S8) конкурирующих cкоростьлимитирующих элементарных стадий.
1.14.КИСЛОТНО-ОСНОВНЫЕ РЕАКЦИИ С ПОЗИЦИЙ ТЕОРИИ МОЛЕКУЛЯРНЫХ ОРБИТАЛЕЙ
1.14.1.«Жесткие» и «мягкие» реагирующие системы
Ранее мы уже познакомились с кислотно-основными реакциями Брёнстеда
A |
|
H + H2O |
|
|
|
A + H3O |
|
|
|
|
|||||
|
|||||||
|
|
|
|
||||
кислота |
основание |
|
продукты |
||||
(отдает |
(присоединяет |
|
реакции |
||||
протон) |
протон) |
|
|
||||
и кислотно-основными реакциями Льюиса
A+  D
D 
 D A
D A
кислота |
основание |
продукт |
(акцептор |
(донор |
реакции |
электронов) |
электронов) |
|
Развивая концепцию Льюиса, теория молекулярных орбиталей определяет кислотно-основные свойства на основе значений энергий занятых МО донора (D) и свободных МО акцептора (A). Наиболее важным в кислотно-
1.14. Кислотно-основные реакции с позиций теории молекулярных орбиталей |
107 |
||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
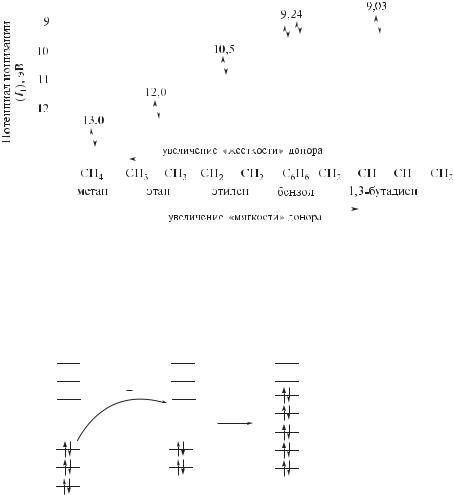
Рис. 1.11. Донорные свойства некоторых органических молекул. Показаны значения первых потенциалов ионизации
основной реакции оказывается переход электронов с ВЗМО донора на НСМО акцептора; эти орбитали ближе других по энергии.
е НСМО
+
ВЗМО
донор D |
акцептор А |
продукт |
(основание) |
(кислота) |
реакции |
Донорные свойства основания (донор D) следует оценивать, таким образом, по уровню энергии его ВЗМО: чем выше энергия ВЗМО (ниже значение I1) донора, тем легче он отдает свои электроны (тем более мягким основанием он является, рис. 1.11).
Акцепторные свойства кислоты (акцептор А) могут быть оценены по уровню энергии его НСМО: чем ниже энергия НСМО акцептора, тем легче он принимает электроны (тем более мягкой кислотой он является, рис. 1.12).
Пользуясь оценками (экспериментальными или расчетными) значений энергий граничных орбиталей реагентов ЕDВЗМО и ЕНСМОА , можно опреде-

108 Глава 1. Природа ковалентной связи. Электронные эффекты. Кислоты и основания
Рис. 1.12. Акцепторные свойства этилена, бензола и 1,3-бутадиена. Показаны значения первого электронного сродства
лить понятия «жесткая» и «мягкая» реагирующие системы. Для этого оценивают разность энергий Δε указанных граничных орбиталей:
Δε = εНСМОА – εDВЗМО.
Если Δε велико, реагирующую систему рассматривают как «жесткую». Если Δε мало, реагирующую систему считают «мягкой».
НСМО
 Δε
Δε
НСМО
ВЗМО |
ВЗМО |
Δε |
|
донор D акцептор А |
донор D акцептор А |
«жесткая» |
«мягкая» |
реагирующая система |
реагирующая система |
1.14.2.Зарядовый и орбитальный контроль органической реакции
С позиций теории МО в рамках метода возмущения молекулярных орбиталей направление и относительная скорость превращения в системе донор–акцеп- тор определяются на основе оценки стабилизирующего изменения полной энергии системы ( Еполн). Чем выше энергия стабилизации системы (Естаб), тем ниже свободная энергия активации ( G≠), тем выше скорость реакции.

1.14. Кислотно-основные реакции с позиций теории молекулярных орбиталей |
109 |
Энергия стабилизации складывается из кулоновского и орбитального членов:
Еполн = Естаб = Екул + Еорб . |
(1) |
Кулоновский член ( Екул) оценивает энергию взаимодействия зарядов на атомах реагентов. Орбитальный член ( Еорб) оценивает энергию взаимодействия занятых i-орбиталей донора и вакантных j-орбиталей акцептора.
Согласно методу возмущения МО, значения энергий Екул и Еорб определяются суммированием соответствующих парных кулоновских и орбитальных взаимодействий между донором D и акцептором A:
|
|
q q |
D A [cD |
cA |
β |
|
]2 |
|
|
E = ∑ μ ν |
+ ∑∑ |
iμ |
jν |
|
μν |
, |
(2) |
||
полн |
μ,ν |
rμνε |
i j |
|
Δε |
|
|
|
|
|
|
|
|
|
|||||
|
|
Eкул |
|
|
Eорб |
|
|
|
|
где qμ — заряды на атомах донора; qν — заряды на атомах акцептора; rμν — расстояние между атомами донора и акцептора; ε — диэлектрическая проницаемость растворителя; сDiμ — значение собственного коэффициента μ-й АО в i-й МО донора D; сjАν — значение собственного коэффициента ν-й АО в j-й МО акцептора А; βμν — резонансный интеграл, оценивающий взаимодействие μ-й и ν-й орбиталей на расстоянии rμν.
В «жестких» реагирующих системах, в которых Δε велико, орбитальный член составляет незначительную величину, а преобладающее значение приобретает кулоновский член:
E ~ E |
кул |
= ∑ qμqν. |
(3) |
|
полн - |
μ,ν rμν |
ε |
|
|
|
|
|
||
Вклад кулоновского члена определяется тем, что ковалентные связи в органических молекулах являются полярными (частично ионными), а в ходе реакции заряды на атомах реагентов могут электростатически взаимодействовать. Стабилизирующий характер кулоновского члена тем выше, чем выше противоположные заряды на атомах донора и акцептора.
Системы, для которых выполняется соотношение (3), подчиняются зарядовому контролю. Согласно методу возмущения МО, выигрыш энергии
от взаимодействия орбиталей реагентов |
Еорб в такой системе крайне мал. |
||||||
В «мягких» реагирующих системах, в которых значение Δε мало или стре- |
|||||||
мится к нулю, вклад орбитального члена |
Еорб резко возрастает: |
||||||
|
|
D |
A [cD cA β |
μν |
]2 |
|
|
E |
~ |
E = ∑ |
∑ |
μ ν |
, |
(4) |
|
Δε |
|
||||||
|
полн - |
орб |
|
|
|
|
|
|
|
|
|
|
|
||
ВЗМО HCMO
где cDμ — значение собственного коэффициента μ-й АО в ВЗМО донора D; cνA— значение собственного коэффициента ν-й АО в НСМО акцептора А.

110 Глава 1. Природа ковалентной связи. Электронные эффекты. Кислоты и основания
Такие системы подчиняются орбитальному контролю. Выражение (4) показывает, что при орбитальном контроле направление реакций и относительные скорости реакций определяются условиями взаимодействия ВЗМО донора и НСМО акцептора, т. е. граничных орбиталей реагентов. Наибольшие эффекты стабилизации достигаются в случае, когда взаимодействие идет по тем атомам, которые имеют максимальные значения коэффициентов cDВЗМО и cHCMOA .
 ОБРАТИТЕ ВНИМАНИЕ!
ОБРАТИТЕ ВНИМАНИЕ!
Реакции, в которых взаимодействие между реагентами определяется зарядами на их атомах, подчиняются зарядовому контролю. Реакции, в которых вза-
имодействие между реагентами определяется энергиями и условиями перекрывания их граничных орбиталей, подчиняются орбитальному контролю.
1.14.3.Концепция граничных орбиталей
Впервые роль граничных орбиталей при взаимодействии органических молекул в 1953 г. обосновал японский химик К. Фукуи в своей концепции граничных орбиталей (Нобелевская премия, 1981 г.):
а) в реакции с электрофильным реагентом наиболее предпочтительно для атаки то положение субстрата, которое имеет максимальное значение собственного коэффициента (т. е. более высокую электронную плотность) в ВЗМО; б) в реакции с нуклеофильным реагентом наиболее предпочтительно для атаки то положение субстрата, которое имеет максимальное значение соб-
ственного коэффициента в НСМО.
Согласно уравнению (1), следует учитывать вклад двух членов в значение Еполн. Однако, как было сказано в разд. 1.14.2, в соответствующих условиях тот или иной член может оказаться преобладающим. Рассмотрим ряд
примеров.
1. Оба реагента несут полные заряды, т. е. являются ионами. В таких реакциях преобладающее значение имеет электростатическое взаимодействие и система подчиняется зарядовому контролю.

 O
O + H
+ H 



 OH pKa 9,98
OH pKa 9,98
CH3CH2 O + H
+ H 
 CH3CH2 OH pKa 15,9
CH3CH2 OH pKa 15,9
2. Реагент заряжен, а субстрат не имеет зарядов. Формулируя концепцию граничных орбиталей, Фукуи подробно рассмотрел относящуюся к данному случаю реакцию электрофильного нитрования аренов.