

Konspekty_Enzimologiya
.pdfвсего, самая доступная), агароза и пр. Целлюлоза и сейчас очень часто используется. У крахмала плохие механические свойства, гель хрупкий, а вот агароза гораздо лучше. Еще в качестве матрицы - запатентованный сефадекс.
Разделение белков идет вблизи изоэлектрической точки, чтобы плотность зарядов была умеренной, иначе не вымыть с колонки.
Матрицы делятся по группам, пришиваемым к ним, на:
-анионообменные матрицы (притягивают -)
-катионообменные матрицы (притягивают +)
Катионообменники:
1)Карбоксилы, пришиваются через метиловые мостики (карбоксиметил). При определении необходимого рН - рКа примерно 4,5. При несоблюдении падает емкость связывания. Предшественникихлорпроизводные. СМ-ионобменник.
2)Сульфогруппы - SO3- через пропил и этил. (сульфопропил и сульфоэтил). Работает и при рН ниже 3. SP- & SEобменники.
3)Фосфат: рКа1=1, рКа2=6.5 => может нести 2 заряда + сложноэфирная связь непрочная + у нас могут быть белки-фосфатазы. Но +: могут специфично связывать белки именно за счет фосфорилирования сахара.
Анионообменники (производные азотсодержащих соединений):
1) Модифицированные амины - I, II, IIIчные.
-DEAE - группы (третичные амины) (диэтиламиноэтил). В щелочных условиях работает плохо, только при рН<7,5.
-QEAE - четвертичный (Q - quartro) амин. К торчащим алкильным группам часто пришивают гидроксилы для избежания неспецифического гидрофобного связывания.
Технические аспекты разделения белков на ионообменниках:
Сначала нужно подобрать подходящую среду, чтобы не шла заметная денатурация при температуре, наличие активаторов и стабилизаторов.
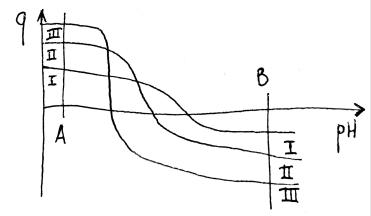
-у белка нет постоянного заряда. Нужно подобрать такой рН, при котором наш белок будет связываться с колонкой, а остальные - нет. В зависимости от рН можно делить и на катионообменниках, и на анионообменниках.
-ионообменная хроматография - обладает высокой емкостью. Слишком большая плотность заряда - плохо, а то белок может прилипнуть навсегда - многоцентровое связывание. Количество центров может меняться в зависимости от условий среды.
На рисунке показано, что при разных рН может быть разная последовательность элюирования разделяемых белков.
Буфер надо брать того же заряда, что и ионобменник (=у него должна быть ионогенная группа, не связывающаяся с обменником иначе изменится рН).
На связывание влияет ионная сила.
Суспендируют в Buf, инкубируют, переносят на колонку. Колонку белком заполняют обычно на треть, максимум на две трети, т.к. насыщение - плохо.
мг/экв - емкость ёмкость ионообменника стандартно указывают по альбумину или гемоглобину.
Весовая единица белка - ориентир для загрузки колонки Неспецифическая сорбция (часто связывается с целлюлозой) - предварительная забивка проводится сывороточным альбумином.
Колонку промывают до почти нулевого уровня выхода белка, т.к. неспецифическое связывание. После разделения нужно быстро довести рН до оптимального для нашего белка.
Элюция градиентом рН (в пределах устойчивости белка и оптимального его существования) - редко используется, так как приходится менять буфер. Элюция градиентом соли - “мешающие ионы”.
Чем выше ионная сила, тем выше время существования белка без связей с носителем. Это определяется концентрацией соли. Разные белки элюируются разной концентрацией соли.
При ионообменной хроматографии на целлюлозе пики немного размываются. Повышение концентрации соли:
1)Ступенчатое изменение - несколько растворов с разной концентрацией - просто и быстро. Но за счет резких скачков - менее эффективное разделение (1
фракция - несколько белков). 2) Непрерывное изменение:
-смешиваем 2 раствора, используя равенства втекающих и вытекающих растворов. Получается экспоненциальная кривая изменения концентрации (колба Бунзена).
-бывает более сложная система: 2 сообщающихся сосуда с растворами разной концентрации, открываем между ними краники. Концентрация возрастает линейно. Если сосуды разные, то получается выгнутая или вогнутая кривая изменения концентрации.
-когда у нас есть сложные смеси, используется более сложное оборудование: 2 сосуда с растворами и многоканальный насос. По определенной программе насос подает определенное количество раствора из 1 и 2 сосудов. Используются level-сенсоры (контроль концентрации белков на выходе). Каждый пик выходит на определенной концентрации соли. Эффективное разделение - до 20 белков.
Плюсы ионообменной хроматографии - большая ёмкость, качество деления мало зависит от параметров колонки.
20.10.15 Гель-фильтрация (проникающая хроматография).
www.ibmc.msk.ru%2Fcontent%2FEducation%2Fw-o_pass%2FMMoB%2F3a.pdf
В колоночной хроматографии важно собрать всю фазу сразу - для этого делают специальное дно => можно повысить площадь сечения колонки => понизить время деления.
Гель-фильтрация
В качестве неподвижной фазы используют гранулы геля с различным диаметром пор. Не подвергаем белок изменениям со стороны среды. Белки в зависимости от размера застревают в разных порах, в разных гранулах.
Чем больше молекулы, тем быстрее они выходят. За счет многократного перехода между гранулами: большие молекулы выходят в объеме V0 (небольшом), маленькие молекулы выходят в объеме, приблизительно равном объему колонки.
Чем больше поры, тем бо́льшие молекулы можно делить.

Более рыхлые гели - для белков, полисахаридов, редко - для НК (для них нужны слишком большие поры в геле, он мнётся).
Матрицы:
-Sephadex - Separating Pharmacea Dextran - декстрановые гели (сшивки - трехуглеродные фрагменты со спиртовой группой посередине -
эпихлоргидрин).
Wiki: Эпихлоргидрин (хлорметилоксиран) — органическое вещество, хлорпроизводное окисипропилена, с формулой СH2-O-CH-CH2Cl.
Эпихлоргидрин получают из пропилена, который хлорируют при температуре 500 °С и давлении 18 атм. до аллилхлорида:
Затем аллилхлорид подвергается действию хлорноватистой кислоты и получают изомерные дихлоргидрины глицерина:
Далее на дихлоргидрины глицерина действуют щелочью (NaOH), в результате чего образуется эпихлоргидрин
Образовавшийся эпихлоргидрин отделяют перегонкой с паром и дистилляцией. Также его можно получить восстановлением хлорированного акролеина.
G 10 (сейчас не используют, изначально - для разделения НМС но это дорого и налипает и задерживает материал) 25, 50, 75, 100, 150, 200.
Маркировка обозначает, сколько мл жидкости впитывает 1 г геля. То есть грамм геля G50 впитает 5 мл, а G150 - 15 мл.
- Изначально гель - в виде сухого порошка; при добавлении воды набухает - надо дать ему время на это. Чем выше значение сефадекс, тем больше воды нужно, тем бо́льшие белки можно разделять (около 750 кДа). Используются колонки тонкие и длинные. G200 - мягкий, это плохо, он сминается. Нужно поддерживать давление, но гель все равно спрессовывается, так как верхние слои давят на нижние. G200 используется крайне редко, почти никогда.
Гели не выкидывают, используют их несколько раз.
-ПАА гели (Bio-gel компании Bio-Rad) Р [число]; бывают от Р2 до Р300). Число обозначает верхний предел молекулярной массы в кДа, т.е. Р4 - до 4000 Да, после которого деление прекращается, и всё выходит в исключённом объёме.
-гели из агарозы (bio-gel A 0,5m-150m; число обозначает миллионы Да) - но эти гели уже довольно хлипкие.
-molselect - ПАА-гели (старые венгерские, производства Reanal, сейчас уже не производятся)
-Sepharose (2B, 4B, 6B; число обозначает процент агарозы в геле; мелкопористые гели) - нельзя автоклавировать, поэтому их стали сшивать: 2BCL (cross-linked), 4B-CL, 6B-CL. Стали обладать очень хорошими механическими свойствами.
-S200-1000 можно было делить в т.ч. рестрикционные фрагменты. Большого применения не нашло. 200-400 нормальные белки, а вот 500-1000 - рестрикты и вирусы.
3основных варианта использования гель-фильтрации:
-разделение НМС и ВМС (G-25 или P-2)
Например, делим сульфат аммония и белок: 1)высаливание 2) смена буфера. Белок выходит в той среде, что был на колонке. Разделение молекул по размерам: все пики в ограниченном объёме => можно вносить только немного материала. Колонки должны быть достаточно узкими, чтобы уменьшить диффузию (а не то пики будут широкими и перекроются). Если хотим много белка: повышаем объем колонки, повышаем концентрацию белка в наносимом растворе, но сокращаем объем (колонка может затормозиться). Рекомендованная концентрация - примерно 10 мг/мл (не больше 20).
V0 - довольно заметная величина (20-25%колонки).
Компромисс: при препаративной очистке берут белков примерно 3-5% колонки. В результате гель-фильтрации пик размывается примерно в 3 раза. В общей сложности можем разделить 5-6 пиков. Но нам обычно важно отделить 1, тот самый, желанный, единственный (а что происходит с остальными пиками, нам всё равно).
На выходе можем отслеживать поглощение. Но активность определяем уже во фракциях. Нужно делать это быстро и точно.
Обычно гель-фильтрация проводится на 1 геле. Но бывает полезно проводить ее и на нескольких, например, сначала разделяем кучу мелких белков от кучи больших белков, а потом хотим смесь больших белков (которая оказалась за гранью разрешающей способности геля по массе белков) разделить между собой - берем другой гель, позволяющий разделять в диапазоне бОльших масс.
Гель-фильтрация даёт возможность определить и другие параметры:
-число компонентов
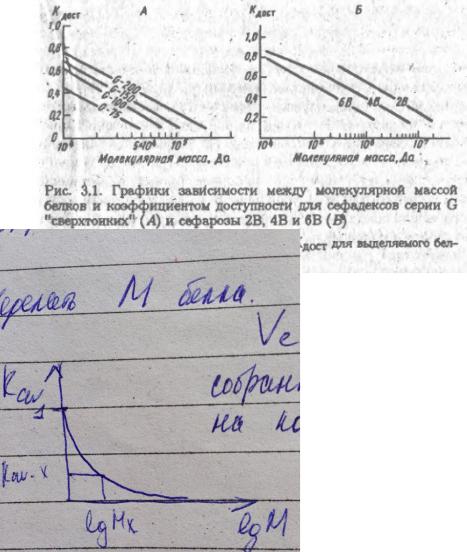
-довольно грубо определить М белка.
Kav = (Ve-Vo)/(Vt-Vo), где
Kav - K available - это коэффициент доступности колонки - условно говоря, это сколько белка может влезть именно в поры гранул.
V0 - объем растворителя между гранулами геля (нулевой объем колонки)
Ve - объем выхода (объем элюента, собранного с момента внесения вещества на колонку до момента выхода его с колонки)
Vt - общий объем колонки.
Строят график зависимости константы доступности от логарифма молекулярной массы. Часто получается выгнутый вниз график, убывающий до 0.
Сейчас М белка обычно определяют электрофорезом, но у него недостаток: с его помощью можно определить размер только полипептида (а белок может состоять из нескольких полипептидных субъединиц). А М нативного белка меряют седиментацией и диффузией в центрифуге.
Основной наш враг - диффузия. Как бороться:
-понижение температуры (холодная комната, холодильная камера для хранения ферментов и фракций, “рубашки” с хладагентами у колонок и малый холодильный шкаф у всего остального)
-малое сечение колонки (Ø/h 0.05)
-равномерность заполнения колонки:
колонку вешают строго вертикально; на дно помещают немного чистого буфера, гель суспендируют в буфере; суспензию заливают в колонку, частицы попадают
на водную подушку. Через некоторое время сверху - чистый буфер. Тогда снизу включается насос => частицы оседают быстрее, когда появляется свободное место - добавляем суспензию. Иногда колонку наращивают насадкой, похожей на воронку, и заливают в неё двойной объём =>колонка забивается за 1 набивку. Поверхность геля не должна нарушаться - накладываем хроматографическую бумажку сверху, поршень с пористым отверстием.
27.10.15 Метод хроматофокусирования. Метод гидрофобной хроматографии белков. Метод аффинной хроматографии. Метод имидазольного радикала. Денатурирующий электрофорез.
При ионообменной хроматографии мы получаем дополнительное концентрирование. Если молекулы имеют заряды, то они взаимодействуют.
А25 поры > G50
При градиенте гель сжимается. Используют ступенчатую элюцию, чтобы этого избежать. Градиент соли можно создать прямо в колонке с гелем. Подаем раствор от менее к более концентрированного.
Не используется, так как нужны большие колонки.
Буферный раствор подбираем так, чтобы не было связывания с ионообменником. Можно наоборот:
сначала связываем буфер с колонкой при определенном pH, потом пропускаем другой буфер - происходит замещение вытеснением. Но при этом происходит смешивание, сдвиг общего рН.
В данном случае ионообменник - также буферная система.
Первая компания Pharmacia:
Хроматофокусирование - метод ионообменной хроматографии, использующий градиент рН, формируемый в слое сорбента в колонке.
1)Степень модификации
2)Ионогенные группы
3)Буфер 1 - полибуфер, задающий градиент
4)Буфер 2 (замещающий) - видимо это и есть типа стартовый
Буферы - амфолины (полиамины, ди- и трикарбоновые кислоты).
Суть хроматофокусирования в том, что мы создаем линейный градиент рН (сначала уравновешивается анионообменник при одном рН, затем наносится полибуфер с начальным рН ниже исходного с постоянной буферной ёмкостью) и вносим белок на колонку. Белок мигрирует (мы его постепенно элюируем, после изготовления полибуфера мы элюируем т.н. стартовым буфером с рН как в начале колонки вроде) до того рН, который соответствует изоэлектрической точке, после чего белок перестает двигаться, т.к. сразу после прохождения изоэлектрической точки приобретает положительный заряд и связывается с катионообменником. Причём стартовый буфер
может постоянно смещать рН и белок вместе с ним до тех пор, пока колонка не кончится.
В pI у белка падает растворимость, помнить о возможности агрегации (у белков в этой точке); добавляем глицерин.
Разница рН 4-7 максимум (различие в 2-3 единицах)
Разделение по pI: если белки не очень отличаются, то разделение затруднено, но если отличаются, то хорошо можно разделить.
Используют чаще после ионообменной хроматографии.
Гидрофобная хроматография
Хроматография + высаливание
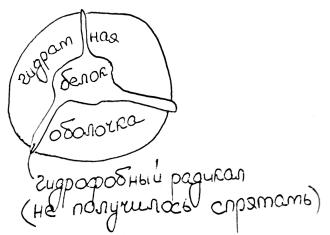
Вода взаимодействует с разными участками белка с разной энергией, которая зависит от гидрофобности аминокислот на поверхности. Если активность воды уменьшается (например, когда мы добавляем соль), вода уходит в первую очередь от наиболее гидрофобных участков, так как нахождение воды там наименее выгодно энергетически. Гидрофобные участки обнажаются, и белки слипаются ими, выпадая в осадок.
Матрица на колонке гидрофильная (это важно) с гидрофобными радикалами. Матрица не может быть полностью гидрофобной, потому что тогда любой белок там сразу развернется, агрегирует и умрет. При некоторой ионной силе раствора происходит слипание гидрофобных групп на носителе и гидрофобных радикалов белка. Но нужны условия, при которых слипание обратимо. Когда понижаем концентрацию соли, понижается сродство белка к носителю, и белок вымывается.
Группы: линейный алкильный радикал, фениловый радикал.
Носитель - в колонку, уравновешиваем раствором, который является растворителем для раствора белка.
Можно разделить не по размеру и не по заряду. Также используют уже почти очищенные белки (после ионообменной).
Чем шире элюция, тем более широкие пики наблюдаем. - у меня написано, что просто при гидрофобной всегда видим широкие пики. Широкая элюцияпроисходит на широких колонках. Ну а гидрофобная широкая потому, что слипание по гидрофобикеочень стохастический процесс, зависящий от многих факторов.
Элюируют повышнием рН, снижением содержания соли, Tween20 или Triton X100.
Аффинная хроматография
2 подхода:
1) Ферменты Основа: белковую молекулу связывают некоторые типа лиганды. Субстрат - аналог.
-Химическая модификация продукта (не очень хорошо)
-Зацепление
2)Белки, связывающие те или иные группы на других белках.
Антитела: +: связывание специфично, устойчиво -: очень легко разрушить при изменении рН.
Для элюции: рН 4-4,5 => после выхода антиген-антитела сразу нейтрализуем. -: надо получать антитела для каждого белка отдельно (дорого и долго). Пришить низкомолекулярный лиганд - в первую очередь. Чтобы избежать превращения при связывании - проводим модификацию субстрата. Используют “ножку”(спейсерная группа) - углеводородную цепочку, на концах - лиганды для
пришивки (глютаровый альдегид). Длина ножки может влиять на сродство фермента к прицепленному лиганду. Длинная ножка - легко схватить. Короткая - сложно. Нужно оценивать, насколько сильно связывание. Если слишком сильное, то плохо, потому что не оторвать фермент. Если слишком слабое, то плохо, потому что держаться совсем не будет.
Как снимать вещество с колонки?
1)Условия, которые изменяют белок. Но изменения должны также легко возвращаться. Например: бацитрацин (с Д-аминокислотами) - сериновые протеазы - изопропиловый спирт (20-30%) и соляная кислота (ее надо быстро нейтрализовать)
2)Можно просто залить много субстрата, который элюирует фермент, конкурируя за него с иммобилизованным аналогом.
3)Можно менять рН или ионную силу. (типа 1М NaCl)
Вбольшинстве случаев белки быстро денатурируют в жестких условиях. Чаще используют: замещение (другим субстратом) или его аналогом (дешевле).
Градация аффинности:
-есть ферменты с абсолютной специфичностью к субстрату
-бывают универсальные (к НАДФ) (групповая специфичность)
Нулевой объем колонки - пропустить что-то большое, водорастворимое - декстран с голубым красителем (большой, в гель не входит - первый разработанный маркер). Иногда не очень большие белки выходили в нулевом объеме - связывание всех белков с активным центром к аденину (с декстраном).
Таким образом, появились специфические носители.
Но многие ферменты все равно имеют сродство, или превращают субстрат; носитель после этого выкидывают.
Матрицу обычно предварительно обрабатывают бромцианом, диэпоксидом, дихлортриазином, трифторэтансульфонилхлоридом, периодатом натрия.
Современный метод имидазольного кольца
В белках есть аминокислота гистидин, имидазольная боковая цепь которой очень любит формировать комплексы с никелем. Если взять четыре гистидина подряд, то такой “хелатор” будет великолепно связывать никель. Можно присоединить шесть гистидиновых кодона в конец трансгена, так что нужный нам белок в бактерии будет содержать такой His-таг. А если снабдить близко расположенными имидазолами гель в
колонке, и залить туда буфер с никелем, то наш белок сформирует “гибридные” комплексы с никелем и с имидазолами геля через свои гистидины. Все остальные белки отмоются. А потом мы берем и элюируем наш белок концентрированным раствором имидазола, который вытесняет имидазольные группы геля из комплексов. Для медицинских целей необходимо отрезать гистидиновый хвост. Аналогично можно использовать кобальт вместо никеля.
Общая тенденция пройденных на этой лекции методов - взять что-то, что вначале легко связывается с белком, а потом так же легко отмывается.
Денатурирующий электрофорез:
SDS- хитрая молекула, это длинный углеводородный хвост и заряженная сульфатная группа. Если добавить SDS к белку, то последний разваливается, и SDS равномерно выстраивается вдоль цепи подобно расческе, при этом число SDS и общий заряд сульфогрупп пропорциональны длине белка. Поэтому белок в комплексе с SDS имеет массу, пропорциональную заряду. В итоге ни та, ни другой на скорость движения в электрическом поле не влияют, и разделение происходит только по длине. При этом каждая новая аминокислота снижает подвижность белка в определенное число раз, и подвижность линейно зависит от логарифма длины белка. Сравнивая подвижности известных и неизвестных белков, можно определять длину неизвестных. Часто думают, что там можно определять молекулярную массу. В общем это неверно, так как масса белка не всегда пропорциональна его длинебывают тяжелые и легкие аминокислоты.
Обычно берется один грамм SDS на 1 грамм белка.
Используют крахмальные колонки :
для отщепления с колонки - фермент узнающий аминоксилотный дополнительный мостик (данный мостик мы встраиваем между матрицей и белком) Бромциансахароза - делаем агарозный гель с бромцианами (для привязки белков, антител). Пришивки: углеводные матрицы - альдегиды, ПАА-гели - гидразин, карбодиимин - при конденсации. (ПОМОГИТЕ РАСШИФРОВАТЬ) о чем это вообще? так лучше?
Видимо, все эти трэшовые реагенты используются для ковалентной иммобилизации разных белков на неподвижном носителе. Но что-то я не въезжаю, к чему это вообще.
3.11.15 Электрофорез. Экстракция белков из геля. Дискэлектрофорез.
Электрофорез.
Раньше в качестве формы для геля использовали трубки. В U-образных трубках, на одном конце был +, на другом -. Белки наносили в центр трубки, там, где изгиб. В результате, белки одного типа двигались с одинаковой скоростью в одну и ту же сторону - получался ряд зон. Раньше зоны детектировали с помощью метода шлиреноптики - искали границы зон с помощью изменения преломления света. Концентрированный белок обычно по-другому преломляет свет. Таким образом было показано, что в сыворотке крови много белков (до 10), а не 2, как считалось раньше.