

2.3 Матеріали, устаткування і методика досліджень
Для проведення досліджень були використані наступні порошки: порошок заліза ПЖ1М1, порошок титану ПТХ7, технічний порошок карбіду бору (ГОСТ 5744 – 74, 50 мкм), лампова сажа П514 (ГОСТ 7885).
Першим етапом роботи є приготування порошкових сумішей, з елементарних порошків Fe, Ti та C (B4C), для систем: Fe – Ti – C та Fe – Ti – B4C, вміст компонентів 20% Fe, 64% Ti та 16% C (B4C), виходячи із співвідношення 20% Fe - 80% TiC, вміст карбіду бору аналогічний до вмісту вуглецю навмисно, хоча вміст вуглецю в системі Fe – Ti – B4C звичайно менший за Fe – Ti – C, (рис. 2.6).
|
|
С, ваг% |
|
Температура, °С |
|
|
|
С, ат% |
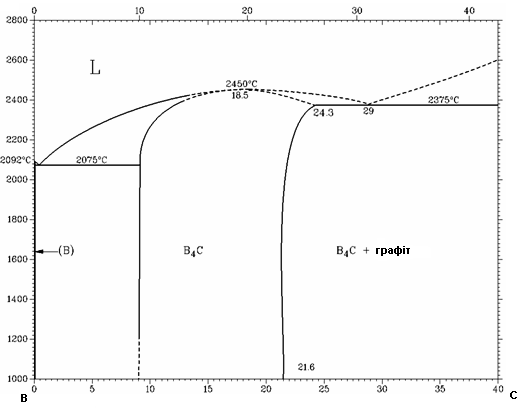
Рисунок 2.6 – Діаграма стану системи В – С (приведена з електронної бібліотеки HRW)
Всі математичні розрахунки тут і в подальшому проводилися з використанням пакету програм Microsoft Office Excel 2007.
Розрахунок наважки шихти для пресування проводиться за формулою:
|
Gn=Vnγср(1-f)k1k2, |
(2.8) |
де: Vn-об´ем пресовки, що визначають по заданому відношенню H/d = 0,5…0,8, d – діаметр пресовки, H – висота пре совки, H – (0,5…0,8)d; а Vn = π d2 H/4; k1 k2 – коефіцієнти, що враховують втрати матеріалу (k1 = 1,005…1,01; k2 – визначають по вмісту кисню у вихідному матеріалі, зв´язуючих речовин, мастил). γk – середня густина багатокомпонентної шихти, що визначаеться за законом адитивності:
|
γср=(γ1 γ2 γ3… γn)/(γ2γ3… γn а1+γ1γ3… γn а2+…γ1γ2 γ3… γn а(n-1)), |
(2.8) |
де: γ1 γ2 γ3 – густини окремих компонентів шихти; а1,а2,а3 – масова частка окремих компонентів шихти.
Розрахунок проводили виходячи з таких вихідних даних, що: d = 30 мм, H = 20 мм, з заданою вихідною пористістю f = 0,2 (20%); приймаючи: k1 = 1,001, k2 = 1.
Отже для суміші системи Fe-Ti-C: γсрFe-Ti-C = 3,7527 г/см3, GFe-Ti-C = 47,6153 г, відповідно:
GFe = 9,5230 г;
GTi = 30,4737 г
GC = 7,6184 г.
А для Fe-Ti-B4C: γсрFe-Ti-B4C = 4,3296 г/см3, Gn Fe-Ti-B4C = 49,1865 г, відповідно:
GFe = 9,8373 г;
GTi = 31,4793 г;
GB4C = 7,8698 г.
Було приготовано шихти, обох складів, що відповідали масі необхідної на три зразка для кожного з вказаних складів.
Змішування проводили в змішувачі типу лабораторному баночному змішувачі з додаванням уайт – аспериту та тіл, що інтенсифікують процес змішування, запобігаючи розшаровуванню порошкових компонентів шихти. Тривалість процесу змішування складає 1,5 год, швидкість обертання 60 об/хв. Після змішування порошкова суміш просушувалась до повного випаровування залишків уайт – аспериту, з неї видалялися єрши. Далі складалися наважки згідно вказаного вище розрахунку.
Пресування заготовок проводилося на гідравлічному пресі ГП – 65, в прес-формах з діаметром робочого каналу 30 мм, при навантаженні 500 МПа. Внутрішні стінки прес-форми підлягали змащування машинним мастилом, для зменшення сил внутрішнього тертя. При пресуванні заготовок використовувалася двостороння схема пресування.
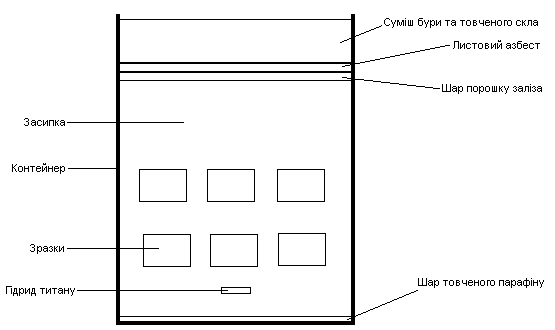
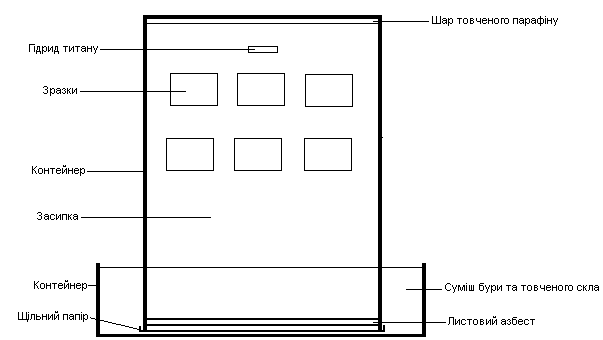
Спікання заготовок в печі з резистивним нагрівом Г-30А, потужністю 30 кВт, без контрольованої атмосфери в активній зоні печі, при температурі 1200 °С, на протязі 1 год. Контроль газового середовища при спіканні заготовок здійснюється за допомогою використання схеми плавкого затвору (рис. 2.7 (а,б)).
Суть схеми полягає у створення контрольованої відновлювальної атмосфери в об’ємі контейнеру, в якому здійснюється спікання зразків.
Зразки поміщаються в циліндричний контейнер, виготовлений з нержавіючої сталі, та запаковуються відповідно до вказаних вище схем:
Невелика кількість парафіну, що поміщується на дно контейнеру необхідна для витіснення кисню повітря з об’єму контейнера, шляхом його розкладання при нагріванні разом з контейнером, за реакцією:
С18Н38 + 27,5 О2 18 СО2↑ +19 Н2О↑
TiH2 при нагріванні до температури 300 – 400 °С розкладається згідно реакції:
TiH2 → Ti + Hатом. ↑
Витісняючи продукти реації розкладу (газифікації парафіну) та утворююче відновлювальне середовище всередині контейнеру, для чого потрібно невелика кількість водню, до того ж атомарній водень має більш високу відновлювальну здатність, що є сприятливим фактом. Гідрид титану повинен бути розміщеним в об’ємі контейнера з великою обережністю, через те що титан, що утворюється при розкладанні TiH2 утворює з залізом легкоплавку евтектику, тому це може призвести до прогорання контейнера.
Роль засипки виконує відпалений пісок, що прокалюється при температурі 1200 °С на протязі 2 год, це дозволяє позбавитись від вологи та по можливості газифікувати небажані домішки, тим самим видаливши їх з об’єму піску.
Шар порошку заліза, це бажаний, але не необхідний компонент паковки. Він необхідний для поглинання можливого залишкового кисню в об’ємі
а)
б)
а) контейнер з плавким затвором зверху;
б) контейнер з плавким затвором знизу.
Рисунок 2.7 – Схема спікання зразків в контейнері з плавким затвором
контейнеру, через свою досить високу спорідненість до кисню; його додавання підвищує вірогідність не окислення зразків.
Прокладка з листового азбесту закладаеться для герметизації робочого об’єму контейнера, затримуючи вихід водню та також перешкоджає затіканню розплаву скла всередину.
Невеликий кількість піску, розміщена шаром над азбестовою прокладкою відіграє роль загущувача розплаву скла, при його плавленні та стіканні в об’єм робочої зони контейнера під своєю вагою, що додаткова забезпечує чистоту робочого об’єму.
Верхній шар паковки складається з суміші порошку скла з додаванням 15 % бури (Na2B4O7*10H2O), або борного ангідриду (B2O3), що дозволяє понизити температуру плавлення скла на 50 °С та сформувати сам так званий плавкий затвор; ступінь якості затвору можна визначити візуально по його зовнішньому вигляду, гарним результатом є наявність рівномірної гладкої (дзеркальної) поверхні затвору, відсутність свищів, каверн на поверхні дзеркала. При необхідності порошок скла може додаватися в процесі спікання прямо в активну зону печі.
Різниці між вказаних різновидів схеми пакування з плавким затвором є, хоча і не суттєва; другий різновид трохи більш трудомісткий, але й більш надійний в плані створення контрольованого газового середовища в об’ємі контейнера, через те що контейнер перевернутий, що додатково перешкоджає виходу водню з робочої зони контейнеру.
Металографічне дослідження:
Вирізка зразків: Вирізка зразків металографічного дослідження проводилася з зразків, що піддавалися спіканню в контейнері з плавким затвором при температурі 1200 °С на протязі 1 год. Зразки попередньо мали розміри 30×(20±2) мм.
Заготовки відбирали холодним способом на метало оброблювальних верстатах. При відборі заготовок, а також при приготуванні зразків були забезпечені всі заходи по запобіганню зразків від нагріву та наклепу (зміцнення металу під дією пластичної деформації), що можуть привести до викривлення структури та зміні властивостей.
При вирізці заготовок холодним способом передбачався припуск від лінії зрізу до краю зразку розміром приблизно 4 мм.
Розмір і форма зразків визначаються задачами дослідження. Зазвичай вирізані зразки циліндричної або прямокутної форми (як в нашому випадку), висота яких рівна 10 мм, а площа досліджуваної поверхні (шліфа) 2-3 см. Для запобігання пошкодження рук, із зразку були зняті задирки та гострі краї обпилені терпугом.
Підготовка зразків до шліфування. Для зручності приготування шліфів отримані зразки розміщувалися в металевих оправках – кільцях круглої форми і заливалися заливалися епоксидним клеєм марки ЭДП (ЭПОК01), представляє собою розчин епоксидної модифікованої смоли з отверджувачем ПЭПА (поліетиленполіамін). Розчин готується з урахуванням кількості отверджувача по відношенню до 100 частин епоксидної смоли 10 – 12 частин отверджувача ПЭПА. Процес приготування клею супроводжується екзотермічною реакцією його тверднення, що обумовлено зшивкою полімерних ланцюгів епоксидної смоли молекулами отверджувача. Час твердіння залежить від величини приготованої проби та температури вихідної композиції (температури зовнішнього середовища). При температурі вище 20 °С, час тверднення скорочується та відповідно навпаки. Клеєва суміш готується безпосередньо перед його використанням. Для точного дозування епоксидної смоли та отверджувача рекомендується. Клейова суміш придатна до використання на протязі 1,5 – 2 годин після приготування. Максимальна міцність, при температурі 20 °С, досягається через 24 години після заливки зразків. При приготуванні можлива зміна кольору клейової суміші, але це не впливає на показники суміші. При змішуванні епоксидної смоли з отверджувачем суміш зазвичай стає жовтуватого кольору. Для інтенсифікації процесу приготування клейової суміші рекомендується підігрівати суміш, використовуючи джерела закритого вогню, до температури не вище 75 – 80 °С, через миттєве спінювання, що робить її непридатною до використання. Підігрівання призводить до зменшення в’язкості клейової суміші, полегшуючи оперування нею. Додавання 15 – 20 % ацетону сприяє зменшенню в’язкості клеєної суміші. Залиті зразки, для прискорення процесу полімеризації, поміщують у сушильну шафу на 1 годину, при температурі не вище 80 °С.
Недолік цього методу в тому що епоксидна клейова суміш та інші подібні речовини, що використовуються для фіксації зразку в оправці, забруднюють поверхню шліфа та обробляючого матеріалу при подальшій обробці.
Шліфування. На торцевій поверхні зразку готується плоска поверхня, з чітко вираженою кромкою без закруглень. Зразок оброблюють на станку з обертаючимся абразивним кругом, моделі 3Г71, при великій швидкості обертання з охолодженням поверхні зразку водою та малим натискуванням. В інакшому випадку, перегрів і сильна механічна дія можуть викликати зміну мікроструктури, що призведе до хибного результату дослідження. Отримана поверхня повинна бути пласкою і не мати завалів. Шліфування металографічних зразків здійснювалося ручним способом.
При ручному способі шліфування на жорстку пласку підкладку (товсте скло або лист металу), що розміщена горизонтально, кладуть наждачний папір. Потім зразок ставлять на наждачний папір заторцьованою площиною і шліфують з легким натискуванням. Шліфування ведуть до повного зникнення рисок, що залишилися після операції торцювання. Коли на шліфі залишаються риски тільки від паперу, шліфування зупиняють. Шліфувальний папір видаляють з підкладки, отряхуючи з неї зерна абразиву, що викришилися і частинки металу. Залишки абразиву видаляють також з підкладки та шліфа, протираючи їх шматком чистої м’якої тканини або ватним тампоном. Після цього операцію шліфування повторюють на папері з більш дрібним зерном. При повторному шліфуванні напрямок руху зразку повинен бути перпендикулярним напрямку рисок, що залишилися від попереднього шліфування (тобто перпендикулярно попередньому напрямку руху зразку). Операцію шліфування повторюють, використовуючи послідовно папір з меншим номером зернистості і кожен раз змінюють напрямок руху шліфа на 90°. Змінення напрямку руху шліфа дозволяє повністю усунути риски, що залишилися після попереднього шліфування. Закінчивши шліфування на папері з найменшою зернистістю, зразок промивають проточною водою і піддають поліровці. При шліфуванні використовувався наждачний папір, що характеризується номером зернистості (пов'язаний з розміром зерен основної фракції абразиву) приведеному в таб. 2.2.
Таблиця 2.2 Крупність основної фракції використовуваного наждачного паперу при шліфуванні шліфів (ГОСТ 3647 – 80)
|
Номер зернистості |
Крупність основної фракції, мкм |
Номер зернистості |
Крупність основної фракції, мкм |
Номер зернистості |
Крупність основної фракції, мкм |
|
200 160 125 100 80 63 50 40 32 25 |
2500 – 2000 2000 – 1600 1600 – 1250 1250 – 1000 1000 – 800 800 – 630 630 – 500 500 – 400 400 – 315 315 – 250 |
20 16 12 10 8 6 5 4 3
|
250 – 200 200 – 160 160 – 125 125 – 100 100 – 80 80 – 63 63 – 50 50 – 40 40 – 28
|
М63 М50 М40 М28 М20 М14 М10 М7 М5
|
63 – 50 50 – 40 40 – 28 28 – 20 20 – 14 14 – 10 10 – 7 7 – 5 5 – 3
|
Полірування. Після завершення шліфування на поверхні шліфа залишаються тонкі риски. Щоб остаточно вирівняти поверхню, шліф полірують до дзеркального блиску. Полірування здійснювалося механічним способом на полірувальному станку, моделі П – 2А. Поліровку здійснювали на дерев’яному диску, змоченому масляною суспензією з алмазним порошком 1 – 2 мкм, при швидкості обертання диску ~80 рад/с. При поліруванні шліф періодично повертали для більш рівномірної обробки всієї поверхні.
Хімічне травлення. Для виявлення мікроструктури отриманого матеріалу застосовувався метод хімічного травлення. При хімічному травленні поверхня шліфа піддається дії хімічних реактивів на протязі певного часу при певній температурі. Травлення зразків носило загальний характер. Травлення проводили при кімнатній температурі поетапно з використанням двох травників. Зважування хімічних речовин для приготування реактивів проводилося на аналітичних терезах. Попереднє травлення здійснювалося травником складу А:
|
пікринова кислота (C6H2(NO2)3OH - тринітрофенол) |
1 г, |
|
HCl |
4 мг, |
|
C2H5OH (розчинник) |
100 мг. |
Даний склад застосовується для виявлення первинної структури при зварюванні або при наплавці вуглецевих та мало вуглецевих сталей.
Микрошліфи спочатку декілька разів травлять складом А по 60 с з проміжними переполіровками до усунення слідів вторинних структур. Після чого промивають проточною водою та сушать етиловим спиртом.
Основне травлення проводять травником складу В:
|
HCl |
4 мг, |
|
CuCl2 |
5 г, |
|
C2H5OH (розчинник) |
25 мг, |
|
Дистильована вода (розчинник) |
30 мг. |
Виявлення мікроструктури матеріалу при хімічному травленні проводилося шляхом змочування поверхні шліфа травниками вказаного складу. При використанні обох травників декілька крапель травника наносилося на поверхню шліфа піпеткою, витримувалося і змивалося етиловим спиртом. Після промивки шліфи ретельно висушували на фільтрувальному папері. Якість травлення перевіряли під мікроскопом, моделі МИМ-7, при тому ж збільшенні, що й дослідження шліфа.
Очищення шліфів проводилося на ультразвуковому диспергаторі, моделі УЗДН – А, в розчині складу 50 % етилового спирту та 50 % бензину, на протязі 5 хв (час промивки визначався експериментально). Після промивки можна візуально спостерігати металічний блиск поверхні мікро шліфа, що зумовлюється очищенням пор зразка від оксидної плівки, забруднень обумовлених процесами шліфовки та поліровки, а також часткове позбавлення від клейову суміш, що відбувається за рахунок високої енергії кавітаційні пухирців.
Мікроскопічне дослідження проводили візуальним спостереженням на металографічному мікроскопі ,МИМ-8 при використанні оптичного збільшення 150 та 600.
Вимірювання мікротвердості. Вимірювання мікротвердості, що дозволяє визначити твердість окремих фаз та структурних складових тонкого поверхневого шару, проводили на приладі моделі ПМТ-3. Випробування проводять згідно ГОСТ 9450 – 76. Мікротвердість визначали на пласкій полірованій чистій поверхні. При приготуванні зразка особлива увага приділялася до запобіганню наклепуванню та нагріванню поверхні, що може призвести до відхилення величини мікротвердості. Дослідження здійснювалось при навантаженні в 50 г, при виборі навантаження керувалися розміром площі ділянки, твердість якої змінюється, і товщиною зразка. В якості індентора використовувалась алмазна піраміда з кутом при вершині 136°. Перед початком випробувань проводилася перевірка приладу на зразках еталонах для встановлення автентичності його результатів.
Щоб не проводити розрахунки мікротвердості за формулою згідно методики, використовували таблицями (додатки до ГОСТ 9450 – 76), в яких приведені значення мікротвердості в залежності від прикладеного навантаження та довжини діагоналі відбитку.
Точність вимірювань при використанні окуляру 10 становить ±0,5 найменшого ділення шкали (при збільшенні об’єктиву 30 – 40×).
Спечені зразки дробилися під прессом з мінімально можливим навантаженням, запобігаючи наклепуванню матеріалу. Отриманий порошок просіювався відділяючи фракцію в 100 мкм; та піддають рентгено – фазовому аналізу разом з шматочками спеченного матеріалу. Рентгенівський фазовий аналіз проводився на дифрактометрі ДРОН-3.0 в кобальтовому Кα-випромінюванні при напрузі U = 30 кВ, струмі І = 17 мА, без фільтру, з використанням фокусування трубки по схемі Брегга-Берттано. Сканування з кроком 0,05° при витримці в точці 4 с. Розрахунок результатів рентгенівського аналізу проводився з використанням програми FindPike, та електронної рентгенівської бібліотеки PCPDFWIN 2000.
Готуютувалися аналогічні за складом до складу лігатури суміші, але їх спікання проводиться в контейнері з плавким затвором, де зразки поміщені в графітову трубу. Наявність графітової труби пояснюється окисленням зразків при попередніх спіканнях; її нагрівання призводить до взаємодії вуглецю труби з повітрям в об’ємі контейнеру, утворюючи відновлювальну атмосферу.
Розмел спеченої композиції проводили у планетарному млині, моделі ВПК2111У2, в середовищі етилового спирту, за режимом:
2,5 хв розмел,
30 хв зупинка та охолодження млина,
2,5 хв розмел.
Отримана емульсія вивантажувалася з барабанів разом з розмеленими тілами в піддони та просушувають на повітрі при кімнатній температурі до повного висихання, випаровування етилового спирту (приблизно на протязі доби). Після чого проводилося зважування отриманого порошку (див. таб. 2.3).
Таблиця 2.3 Втрати маси на операції розмелу порошкових сумішей
|
Склад порошкової суміші |
Маса до розмелу, г |
Маса після розмелу, г |
|
Fe – Ti – C |
150 |
136,20 |
|
Fe – Ti – B4C |
150 |
145,85 |