

1.5.2. Расчет термодинамических функций сольватации методами компьютерного моделирования
В методах молекулярной динамики (МД) и Монте Карло (МК) взаимодействия между частицами в системе описываются различными потенциалами. Параметры этих потенциалов могут быть получены из эксперимента, например, жесткость связи рассчитывают на основе силовых констант по данным ИК-спектров, либо из ab initio квантовохимических расчетов (геометрия, заряды), либо могут подбираться так, чтобы симуляции как можно лучше воспроизводили различные свойства реальных веществ: плотность, температуру, энтальпию испарения, коэффициенты диффузии.
В методе Монте-Карло (алгоритм Метрополиса) на каждом шаге генерируются небольшие случайные изменения координат частиц, при этом вероятность принятия этих изменений пропорциональна еxp(–ΔE/kT), если ΔE>0, и 1, если ΔE<0. Если шаг принят, то новая конфигурация добавляется к фазовой траектории системы. Метод молекулярной динамики основан на решении уравнений классической механики на небольших (1-2 фс) отрезках времени, исходя из известных потенциалов взаимодействий и случайно сгенерированных начальных скоростей всех частиц. Он часто применяется для изучения фолдинга белков, диффузионных процессов. Используя оба метода, можно напрямую изучать состав окружения каждой молекулы в растворе, предсказывать вид функций радиального распределения, рассчитывать на основании тех или иных критериев число водородных связей между различными молекулами.
Эти методы могут применяться и для расчета термодинамических функций сольватации. Теоретически для расчета энтальпии сольватации мы бы могли усреднить значение энтальпии системы с растворенным веществом и без него по достаточно длинным траекториям, однако на самом деле флуктуации этих значений намного превысят величины энтальпий сольватации. Вычисление же энергии Гиббса подобным способом принципиально невозможно. Поэтому прибегают к использованию таких методов, как метод возмущений свободной энергии (FEP) и термодинамическое интегрирование (TI).
В методе FEP переход от
начального состояния λ = 0 к конечному
λ = 1 трактуется как серия дискретных
изменений потенциалов взаимодействий.
Например, для описания сольватации в
состоянии λ = 0 растворенное вещество
не взаимодействует с растворителем, а
в последующих состояниях величины
потенциалов всех невалентных взаимодействий
между растворенным веществом и
растворителем могут составлять 10%, 20%,
30% и т.д. от полного потенциала, достигая
100% в состоянии λ = 1. Для каждого из
состояний проводится отдельная симуляция,
на каждом шаге которой рассчитывается
не только энергия при текущем значении
параметра λ
![]() ,
но и энергия при потенциале, соответствующем
следующему шагу
,
но и энергия при потенциале, соответствующем
следующему шагу
![]() .
Энергия Гиббса при переходе системы от
начального состояния к конечному
рассчитывается по уравнению:
.
Энергия Гиббса при переходе системы от
начального состояния к конечному
рассчитывается по уравнению:
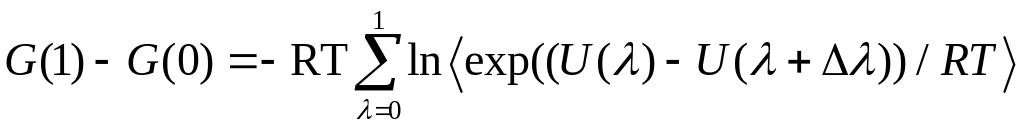 .
(29)
.
(29)
Точность расчета тем выше, чем меньше шаг изменения λ.
В методе термодинамического интегрирования переход к конечному состоянию разбивается на шаги аналогичным образом, а изменение энергии Гиббса рассчитывается по формуле:
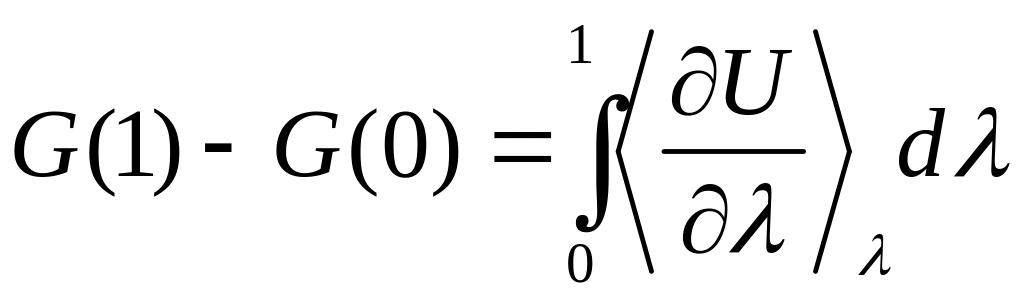 .
(30)
.
(30)
Применение этих методов дает хорошо согласующиеся с экспериментальными данными энергии Гиббса гидратации различных алканов и спиртов [67, 68]. Для расчета энтальпии сольватации можно использовать температурную значимость энергии Гиббса от температуры (ур-е 20), однако ошибка этого расчета всегда будет намного выше, чем для энергии Гиббса.
Большой интерес для расчета термодинамических функций сольватации одноатомных молекул и частиц с заданным произвольным потенциалом взаимодействия (в частности, жестких сфер) представляет метод вставки частиц Видома [69]. Для каждой конфигурации из траектории симуляции растворителя производится большое число вставок изучаемой частицы в случайно выбранные точки пространства, при этом рассчитывается изменение потенциальной энергии. Для нахождения избыточного по сравнению с идеальной газовой фазой химического потенциала проводится усреднение по всем вставкам во все конфигурации. В случае NPT-ансамбля формула расчета имеет вид:
![]() ,
(31)
,
(31)
где ΔU – изменение потенциальной энергии при каждой вставке частицы, V – объем системы. Точность метода зависит от числа вставок и длины траектории, результаты хорошо воспроизводятся уже при 105 вставок на каждую конфигурацию. Величины, полученные этим методом для сольватации инертных газов и метана (в модели с объединенными атомами углерода и водорода) в воде, хорошо воспроизводят эксперимент [70] При этом вычислительные затраты гораздо меньше, чем в методах пертурбации свободной энергии и термодинамического интегрирования. Метод, однако, неприменим для частиц слишком большого размера, например, для расчета энергий образования полостей большого размера из-за ничтожной вероятности их появления в жидкости за время симуляции, а также для многоатомных молекул.