

31. Мобилизация липидов из жировых депо
Состав липопротеинов. Липопротеины состоят из ядра, в котором находятся триацилглицеролы (ТАГ), эфиры холестерола (ЭХ), и поверхностного монослоя из фосфолипидов (ФЛ), свободного или неэстерифицированного холестерола (СХ) и апопротеинов. Функцией липопротеинов является транспорт липидов. Без этой транспортной формы липиды были бы нерастворимы в плазме крови.
Синтез хиломикронов (ХМ). В энтероцитах идет эстерификация 2-МАГ и ХС жирными кислотами (ЖК), образуются ТАГ и ЭХ, из которых затем формируются ХМ. Всосавшиеся ЖК активируются, преобразуясь в ацил-КоА. Это происходит в гладком эндоплазматическом ретикулуме. Важнейшим структурным компонентом ХМ является белок (апо В-48). В составе одной частицы ХМ находится одна молекула апо В-48.
ХМ секретируются с базолатеральной поверхности клеток кишечника в лимфу, а оттуда через грудной лимфатический проток попадают в систему кровообращения. После того как ХМ попадают в лимфу, они получают от ЛПВП апо С-II, С-III и апо Е.
Катаболизм ХМ. Попадая в систему кровообращения, ХМ быстро подвергаются катаболизму. Уровень ТАГ в плазме крови возрастает через 2 ч после приема пищи, а через 4 ч начинает постепенно снижаться. Время разрушения ХМ зависит от гидролиза ТАГ под действием липопротеинлипазы (ЛПЛ). Кофактором этого фермента является апо С-II. Гидролиз ТАГ приводит к уменьшению размеров ХМ, образуется избыточное количество поверхностных элементов по отношению к объему частиц.
Остатки ХМ разрушаются в печени. Таким образом, в процессе своего катаболизма в кровотоке ХМ поставляют ЖК клеткам периферических тканей (жировой и мышечной), в то время как ХС пищи попадает в печень.
Обмен липопротеинов очень низкой плотности (ЛПОНП), липопротеинов промежуточной плотности (ЛППП) и липопротеинов низкой плотности (ЛПНП). Основной функцией этих липопротеинов является транспорт жирных кислот в составе ТАГ из печени к периферическим тканям, особенно в жировую и мышечную. Для синтеза ЛПОНП в гепатоцитах требуется белок апо В-100 и липиды ЭХ, ТАГ и ФЛ.
Триацилглицеролы для ЛПОНП синтезируются путем эстерификации глицерола жирнымих кислотами. Жирные кислоты поступают в гепатоциты из плазмы крови (источником их является, например, липолиз в жировой ткани) или синтезируются de novo в печени. Образование ЛПОНП регулируется за счет контроля синтеза апо В-100.
Новосинтезированная частица ЛПОНП содержит одну молекулу белка — апо В-100. Другие белковые компоненты (апо С-II, апо С-III и апо Е) поступают на неё от ЛПВП после того, как ЛПОНП попадают в плазму крови. Они требуются для ускорения метаболизма ЛПОНП.
Обмен ЛПОНП. На эндотелии сосудистой стенки ТАГ в составе ЛПОНП подвергаются действию фермента ЛПЛ. Необходимым кофактором для проявления активности ЛПЛ является апо С-II. ЛПЛ образуется в клетках многих тканей, среди которых наибольшее значение имеют жировая ткань, скелетная и сердечная мышцы, молочная железа во время лактации. ЛПЛ катализирует гидролиз ТАГ в составе ХМ и ЛПОНП до жирных кислот, моноацилглицеролов (МАГ), в результате ЛПОНП превращаются в кровотоке в ЛППП. Фермент проявляет слабую активность по отношению к МАГ и ФЛ.
В жировой ткани синтез ЛПЛ стимулирует инсулин. Тем самым обеспечивается поступление жирных кислот в адипоциты для синтеза и хранения в виде ТАГ. В мышцах ЛПЛ позволяет использовать жирные кислоты для окисления в периоды между приемами пищи, а инсулин подавляет образование этого фермента.
Липопротеины промежуточной плотности (ЛППП). Образование ЛППП происходит из ЛПОНП. Около 75 % ЛППП попадает в печень после связывания апо Е с соответствующими рецепторами. В печени они полностью разрушаются. Около 25 % ЛППП в кровотоке подвергается действию другого липолитического фермента, печеночной липазы (ПЛ). Этот фермент катализирует дальнейшее расщепление ТАГ в составе ЛППП. В результате ЛППП превращаются в ЛПНП.
Синтез триацилглицеролов.Клетки большинства тканей, особенно печени и жировой ткани, обладают способностью накапливать ТАГ. Жировая ткань функционально специализируется на хранении (запасании) и мобилизации ТАГ (рис. 12.1).
Предшественниками для синтеза ТАГ являются глицерол-3-фосфат и активированные жирные кислоты. В печени глицерол-3-фосфат может образовываться или в результате фосфорилирования глицерола, или из глюкозы как промежуточный продукт гликолиза. В жировой ткани единственным источником образования глицерол-3-фосфата является гликолиз.
Вслед за перевариванием пищи в плазме крови увеличивается концентрация глюкозы, инсулина, липопротеинов, богатых ТАГ. Стимулируется активность ЛПЛ для гидролиза ТАГ в составе липопротеинов, и снижается активность жиромобилизующей липазы в жировой ткани. Наряду с этим стимулируется образование ТАГ в жировой ткани. Натощак или при повышенной потребности в энергии во время физической работы, повышении уровня катехоламинов, гормона роста, АКТГ и глюкагона в плазме крови, снижении секреции инсулина эти процессы меняются на противоположные — увеличивается липолиз в жировой ткани и высвобождаются жирные кислоты. Они используются в качестве источника энергии. Глицерол используется для глюконеогенеза.

Рис. 12.1. Депонирование и расщепление нейтрального жира в адипоцитах
Липопротеины
низкой плотности (ЛПНП). ЛПНП
удаляются из кровотока путем взаимодействия
с рецепторами для ЛПНП (другое их название
апо В/Е рецепторы)
(рис. 12.2). Доля
этого процесса в удалении всех ЛПНП
составляет 75 %. Остальная часть удаляется
с помощью «мусорных» рецепторов
макрофагов, имеющих низкую способность
связывания. Этот путь получил образное
название «мусорный пут
Рис. 12.2. Поступление ЛПНП в клетки и их внутриклеточный катаболизм
После связывания ЛПНП комплекс рецептор — ЛПНП переносится в клетку посредством эндоцитоза; затем он сливается с лизосомами и разрушается. Внутриклеточное высвобождение холестерола, происходящее таким путем, вызывает следующие эффекты: а) снижает синтез ключевого фермента образования собственного, клеточного, холестерола — гидроксиметилглютарил-КоА (ГМГ-КоА) редуктазы; б) снижает синтез рецепторов для ЛПНП; в) активирует фермент ацетил-КоА-холестерол ацилтрансферазу (АХАТ), который катализирует образование из метаболически активной формы — СХ — метаболически неактивной формы — ЭХ (рис. 12.3).

Рис. 12.3. реакция, катализируемая АХАТ
В отличие от регуляторного действия рецепторов к ЛПНП на обмен холестерола в клетках, мусорные рецепторы продолжают транспортировать ХС в клетку без торможения по принципу обратной связи. Тем самым макрофаги превращаются в пенистые клетки. Их образование рассматривается как начальный этап атеросклероза.
Метаболизм липопротеинов высокой плотности (ЛПВП).ЛПВП синтезируются в клетках печени и кишечника. Сразу после секреции ЛПВП имеют вид плоских дисков, содержащих ФЛ. Белковым компонентом их является апо А. Из тканей и клеточных мембран на них поступает холестерол. Под действием фермента лецитин-холестерол ацилтрансферазы (ЛХАТ) из СХ и жирной кислоты фосфатидилхолина образуются ЭХ. В результате частицы ЛПВП созревают, принимая форму глобулы. Затем ЭХ транспортируются на ЛППП, ЛПНП, обломки ХМ с помощью липидпереносящего белка (ЛПБ) или апо D.
32. Окисление глицерола.
Окисление глицерола в клетках.
Начинается оно с активирования этого вещества:
З-фосфоглицериновый альдегид подвергается дальнейшему окислению с участием ферментов, катализирующих реакции второго этапа гликолиза. При аэробном окислении одной молекулы глицерола генерируется 23 молекулы АТФ:
3-глицеролфосфатдегидрогеназная реакция – 3 АТФ,
3-фосфоглицеральдегиддегидрогеназная реакция – 6 АТФ,
фосфоглицераткиназная реакция – 2 АТФ,
пируваткиназная реакция – 2 АТФ,
пируватдегидрогеназная реакция – 6 АТФ,
окисление ацетил-КоА в цикле Кребса – 12 АТФ.
Поскольку 1 молекула АТФ затрачивается в глицеролкиназной реакции, то итоговый энергетический эффект всего процесса – 22 молекулы АТФ.
β-кисление жирных кислот в клетках
С
наибольшей интенсивностью этот процесс
протекает в печени, где жирные кислоты
окисляются до ацетил-КоА. В сердце и
других органах они способны окисляться
до углекислого газа и воды. Протекает
β-окисление жирных кислот в пространстве
между внутренней и наружной мембранами
митохондрий. Начинается оно с активирования
молекулы жирной кислоты:
Так завершается один цикл β-окисления жирной кислоты, во время которого генерируется по одной молекуле ФАД-Н2 и НАД-Н2, окисляемых в дыхательной цепи митохондрий, и ацетил-КоА, способного окисляться в цикле Кребса.
Укоротившийся на 2 углеродных фрагмента активированный остаток пальмитиновой кислоты (ацил-КоА) таким же образом, цикл за циклом, подвергается дальнейшему β-окислению. На последнем этапе этого процесса образуется активная форма масляной кислоты (бутирил-КоА). Она также подвергается β-окислениию:

Из этого четырехуглеродной активированной жирной кислоты генерируется 2 молекулы ацетил- КоА, но не по две, а по одной молекуле ФАД-Н2 и НАД-Н2(образовавшаяся молекула ацетил- КоА не подвергается дальнейшему β-окислению).
Энергетический эффект β-окисления жирных кислот(Х)рассчитывается по формуле: Х = n/2 X 17 – (5-1), где n – число углеродных атомов, включая группу –СООН; 17 – число молекул АТФ, генерируемых во время одного цикла: при окислении ФАД-Н2 (2 АТФ),НАД-Н2 (3 АТФ) и ацетил-КоА в цикле Кребса.(12 АТФ); 5 – количество АТФ, не образовашегося на последнем этапе β-окисления; 1 – количество АТФ, затрачиваемое на активацию жирной кислоты (образование ацил-КоА).
β-окисления жирных кислот в печени.
В этом органе основная масса жирных кислот окисляется до ацетил-КоА. Две молекулы последнего последнего соединяются между собой в результате реакции, катализируемой тиолазой. Образовавшаяся ацетоуксусная кислота током крови доставляется в мышечную и другие ткани, где обратно расщепляется до ацетил-КоА, который окисляется в цикле Кребса. Часть молекул ацетоуксусной кислоты, восстанавливаясь ионами водорода, отщепляемыми от НАД-Н2, превращается в β-гидрооксимасляную килоту. Какое-то количество молекул ацетоуксусной кислоты, отщепляя от карбоксильной группы углекислый газ, превращается в ацетон. Общее количество ацетоуксусной, β-гидрооксимасляной кислот и ацетона называют кетоновыми телами. Количество последних в тканях увеличивается (кетоз) при усиленном образовании их и недостаточно эффективном окислении.
Окисление пропионовой кислоты,образовавшейся в рубце при расщеплении микроорганизмами клетчатки.
Сукцинил-КоА в дальнейшем окисляется в цикле Кребса.
33. β - окисление ЖК
Триацилглицерины поэтапно расщепляется тканевыми липазами.
![]()
Ключевым ферментом липолиза является гормональнозависимая ТАГ-липаза. Образующиеся на этом этапе распада жиров глицерин и жирные кислоты окисляются в тканях с образованием энергии.
Различают несколько вариантов окисления жирных кислот: α - окисление, β - окисление, ω - окисление. Основным вариантом окисления жирных кислот является β - окисление. Оно наиболее активно протекает в жировой ткани, печени, почках и сердечной мышце.
β - окисление заключается в постепенном отщеплении от жирной кислоты двух углеродных атомов в виде ацетил - КоА с освобождением энергии. Запас жирных кислот в клетках сосредоточен в цитозоле, где протекает активация жирных кислот с образованием ацил - КоА

Сам процесс β-окисления ацил-КоА происходит в митохондриях. Митохондриальная мембрана непроницаема для длинноцепочечных ацил - КоА. В переносе их внутрь митохондрий участвует специальный переносчик карнитин (метил, гидроксипроизводное аминомасляной кислоты). Ацил - КоА образует с карнитином комплекс, который после переноса жирной кислоты внутрь митохондрий распадается.

Энергетическая эффективность бета - окисления жирных кислот складывается из энергии окисления ацетил - КоА в цикле Кребса и энергии, освобождающейся в самом бета – цикле. Энергия окисления жирной кислоты тем выше, чем длиннее её углеродная цепь. Количество молекул ацетил - КоА из насыщенной жирной кислоты и количество образующихся из них молекул АТФ определяется по формулам:
n=N/2, где n-количество молекул ацетил - КоА, N- число атомов углерода в жирной кислоте.
Количество молекул АТФ за счёт окисления молекул ацетил-КоА = (N/2)*12
Число β - циклов окисления на один меньше, чем количество образующихся молекул ацетил-КоА, поскольку в последнем цикле масляная кислота за один цикл переходит в две молекулы ацетил-КоА, и рассчитывается по формуле
Количество β - циклов = (N/2)-1
Количество молекул АТФ в β - цикле рассчитывается, исходя из последующего окисления образовавшихся в нём НАДН2 (3 АТФ) и ФАДН2 (2 АТФ) по формуле
Количество молекул АТФ, образующихся в β - циклах = ((N/2)-1)*5
2 макроэргические связи АТФ расходуются на активацию жирной кислоты
Суммарная формула для подсчёта выхода АТФ при окислении насыщенной жирной кислоты имеет вид: 17(N/2)-7.
Окисление ненасыщенных жирных кислот на начальных стадиях представляет обычное β - окисление до места двойной связи. Если эта двойная связь находится в β - положении, то продолжается окисление жирной кислоты со второго этапа (минуя стадию восстановления ФАД→ ФАДН2). Если двойная связь находится не β - положении, то ферментами еноилтрансферазами связь перемещается в β – положение. Таким образом, при окислении ненасыщенных жирных кислот образуется меньше энергии (теряется образование ФАДН2 на каждую двойную связь). Она рассчитывается по формуле:
7(N/2)-7-2m, где m-число двойных связей.
Химизм β - окисления насыщенных жирных кислот

34. Этапы биосинтеза высших жирных кислот, характеристика синтазы высших жирных кислот.
Высшие жирные кислоты могут быть синтезированы в организме из метаболитов углеводного обмена. Исходным соединением для этого биосинтеза является ацетил-КоА, образующийся в митохондриях из пирувата – продукта гликолитического распада глюкозы. Место синтеза жирных кислот – цитоплазма клеток, где имеется мультиферментный комплекссинтетаза высших жирных кислот. Этот комплекс состоит из шести ферментов, связанных с ацилпереносящим белком, который содержит две свободные SH-группы (АПБ-SH). Синтез происходит путём полимеризации двууглеродных фрагментов, конечным продуктом его является пальмитиновая кислота – насыщенная жирная кислота, содержащая 16 атомов углерода. Обязательными компонентами, участвующими в синтезе, являются НАДФН (кофермент, образующийся в реакциях пентозофосфатного пути окисления углеводов) и АТФ.
Ацетил-КоА поступает из митохондрий в цитоплазму при помощи цитратного механизма (рисунок 20.1). В митохондриях ацетил-КоА взаимодействует с оксалоацетатом (фермент –цитратсинтаза), образующийся цитрат переносится через митохондриальную мембрану при помощи специальной транспортной системы. В цитоплазме цитрат реагирует с HS-КоА и АТФ, вновь распадаясь на ацетил-КоА и оксалоацетат (фермент – цитратлиаза).

Рисунок 20.1. Перенос ацетильных групп из митохондрий в цитоплазму.
Начальной реакцией синтеза жирных кислот является карбоксилирование ацетил-КоА с образованием малонил-КоА (рисунок 20.2). Фермент ацетил-КоА-карбоксилаза активируется цитратом и ингибируется КоА-производными высших жирных кислот.

Рисунок 20.2. Реакция карбоксилирования ацетил-КоА.
Затем ацетил-КоА и малонил-КоА взаимодействуют с SH-группами ацилпереносящего белка (рисунок 20.3).

Рисунок 20.3. Взаимодействие ацетил-КоА и малонил-КоА с ацилпереносящим белком.
Далее происходит их конденсация, декарбоксилирование и восстановление образовавшегося продукта (рисунок 20.4).
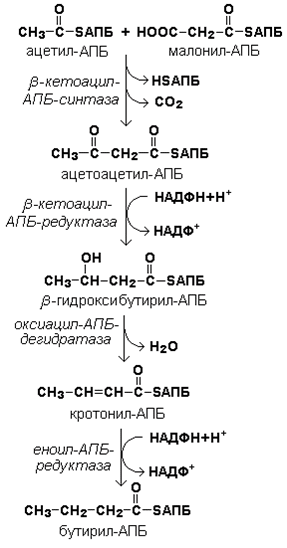
Рисунок 20.4. Реакции одного цикла биосинтеза жирных кислот.
Продукт реакции взаимодействует с новой молекулой малонил-КоА и цикл многократно повторяется вплоть до образования остатка пальмитиновой кислоты.
Запомните основные особенности биосинтеза жирных кислот по сравнению с β-окислением:
-
синтез жирных кислот в основном осуществляется в цитоплазме клетки, а окисление – в митохондриях;
-
участие в процессе связывания СО2 с ацетил-КоА;
-
в синтезе жирных кислот принимает участие ацилпереносящий белок, а в окислении – коэнзим А;
-
для биосинтеза жирных кислот необходимы окислительно-восстановительные коферменты НАДФН, а для β-окисления – НАД+ и ФАД.
35.
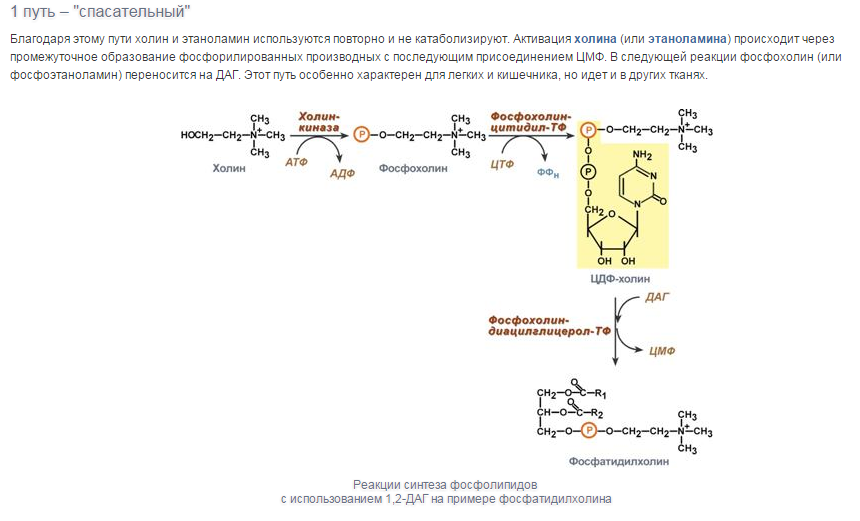
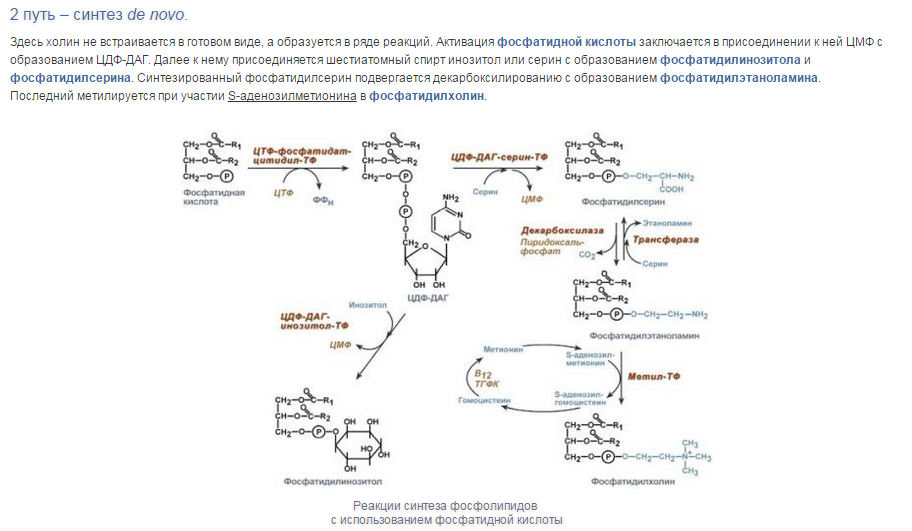
Уравнения реакций синтеза триацилглицеролов
и фосфатидов: фосфатидилхолина…

36. Биологическая роль холестерола. Этапы синтеза холестерола и его транспорт по крови.
Исходным соединением для синтеза холестерола является ацетил-КоА (см. 20.2.3, рисунок 20.6). Ферменты, катализирующие реакции синтеза, содержатся в цитоплазме и эндоплазматическом ретикулуме многих клеток. Наиболее активно этот процесс происходит в печени. В организме человека в сутки синтезируется около одного грамма холестерола.
21.1.2. Биосинтез холестерола включает три основные стадии (рисунок 21.1).

Рисунок 21.1 Синтез холестерола.
На первой стадии образуется мевалоновая кислота (рисунок 21.5, а).
На второй стадии мевалоновая кислота превращается в изопентенилпирофосфат («активный изопрен»), 6 молекул которого конденсируются в сквален (рисунок 21.5, б).
На третьей стадии сквален превращается в холестерол (рисунок 21.5, в).
Всего для синтеза 1 молекулы холестерола используется 18 молекул ацетил-КоА: для образования «активного изопрена» требуется 3 молекулы; в последующих реакциях конденсации участвуют 6 молекул «активного изопрена»; 3 × 6 = 18.
21.1.3. Скорость синтеза холестерола в организме регулируется по механизму отрицательной обратной связи (рисунок 21.5, пунктирная стрелка). Фермент β-гидрокси-β-метилглутарил-КоА-редуктаза катализирует лимитирующую реакцию биосинтеза холестерола. Холестерол является корепрессором синтеза данного ферментного белка, что приводит к снижению скорости катализируемой реакции. Поэтому при поступлении избытка холестерола с пищей синтез эндогенного холестерола прекращается.
Холестерол является компонентом биологических мембран, из него в организме образуются стероидные гормоны, витамин D3, желчные кислоты (см. 14.1.2). Избыток холестерола превращается в печени в желчные кислоты (см. 20.1.3, рисунок 20.3), а также выделяется с желчью в кишечник и выводится с калом.
21.1.4. Нормальное содержание холестерола в сыворотке крови человека составляет 3,9 – 6,3 ммоль/л. Транспортной формой холестерола в крови являются липопротеины (см. далее 21.4.2). Если нарушается соотношение между поступлением холестерола в организм и его выведением, то содержание холестерола в тканях и крови изменяется. Повышение концентрации холестерола в крови (гиперхолестеролемия) может приводить к развитию атеросклероза и желчно-каменной болезни.
37. Синтез и использование кетоновых тел. Понятие и причины кетонемии и кетонурии.
РЕАКЦИИ СИНТЕЗА КЕТОНОВЫХ ТЕЛ
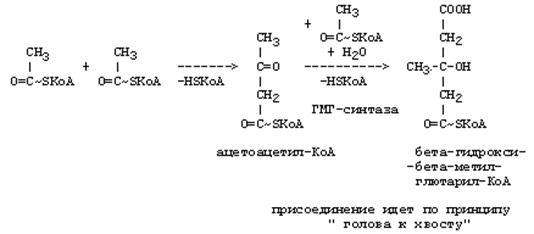
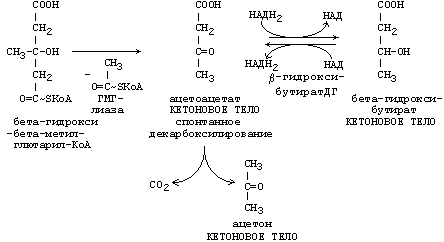
Ацетон, который образуется при спонтанном (неферментативном) декарбоксилировании ацетоацетата, в организме не используется. Он выводится с выдыхаемым воздухом, секретом потовых желёз и мочой. В норме концентрация ацетона в крови мала и обычными реакциями не определяется.
Кетоновые тела синтезируются в печени, легко проходят через митохондриальные и клеточные мембраны и поступают в кровь. Кровью они транспортируются во все другие ткани. Используются только ацетоацетат и бета-гидроксибутират.
Происходит в митохондриях (кроме клеток печени).
Бета-гидроксибутират превращается в ацетоацетат, а ацетоацетат вступает в реакцию с промежуточным продуктом ЦТК - сукцинил-КоА.

Пути использования образовавшегося из кетоновых тел АцетилКоА зависят от функционального состояния клетки (энергетический заряд) и ее специфики.
В ткани, которая получила этот Ацетил-КоА, он может быть использован для разных целей, но чаще всего в ЦТК для получения энергии.
В норме процессы синтеза и использования кетоновых тел уравновешены, поэтому концентрация кетоновых тел в крови и в тканях обычно очень низка, и составляет 0.12 - 0.30 ммоль/л.
Однако при общем или при углеводном голодании может нарушаться баланс между образованием и утилизацией кетоновых тел. Это связано с тем, что скорость образования кетоновых тел зависит от скорости b-окисления жирных кислот в печени, а процесс b-окисления ускоряется при усилении липолиза (распада жира) в жировой ткани. Усиление липолиза может происходить под действием гормона адреналина, при мышечной работе, при голодании. При недостатке инсулина (сахарный диабет) также происходит усиление липолиза. При усилении липолиза увеличивается скорость утилизации кетоновых тел, которые являются важными источниками энергии при мышечной работе, голодании.
Постепенное истощение запасов углеводов при сахарном диабете приводит к относительному отставанию утилизации кетоновых тел от кетогенеза. Причина отставания: не хватает сукцинил-КоА и ЩУК, которые, в основном, являются продуктом обмена углеводов. Поэтому верно выражение: "Жиры сгорают в пламени углеводов". Это означает, что для эффективного использования продуктов распада жира необходимы продукты углеводного обмена: сукцинил-КоА и ЩУК.
Таким образом, при углеводном голодании концентрация кетоновых тел в крови увеличивается. На 3-й день голодания концентрация кетоновых тел в крови будет примерно 2 - 3 ммоль/л, а при дальнейшем голодании - гораздо более высокой. Это состояние называют ГИПЕРКЕТОНЕМИЯ. У здоровых людей при мышечной работе и при голодании наблюдается гиперкетонемия, но она незначительна.
Похожая ситуация характерна для САХАРНОГО ДИАБЕТА. При сахарном диабете клетки постоянное сильнейшее углеводное голодание, потому что глюкоза плохо проникает в клетки. Наблюдается активация липолиза и повышается образование кетоновых тел. При тяжелых формах сахарного диабета концентрация кетоновых тел в крови может быть еще выше, и достигать опасных для жизни значений: до 20 ммоль/л и более.
Почему же накопление кетоновых тел является опасным для организма?
Все кетоновые тела являются органическими кислотами. Их накопление приводит к сдвигу pH в кислую сторону. В клинике повышение концентрации кетоновых тел в крови называется "ГИПЕРКЕТОНЕМИЯ", а сдвиг pH при этом в кислую сторону - "КЕТОАЦИДОЗ". Нарушается работа многих ферментативных систем. Увеличение концентрации ацетоацетата приводит к ускоренному образованию ацетона. Ацетон - токсичное вещество (органический растворитель). Он растворяется в липидных компонентах клеточных мембран и дезорганизует их. Страдают все ткани организма, а больше всего - клетки нервной ткани. Это может проявляться потерей сознания (ГИПЕРГЛИКЕМИЧЕСКАЯ КОМА). В очень тяжелых случаях может наступить гибель организма. Организм пытается защититься, поэтому часть кетоновых тел удаляется с мочой. Появление кетоновых тел в моче - это КЕТОНУРИЯ. Для распознавания гипер- и гипогликемической комы применяется экспресс-диагностика кетоновых тел. Основана на том, что гиперкетонемия приводит к выведению кетоновых тел с мочой (кетонурия). Поэтому проводят цветную реакцию на наличие кетоновых тел в моче. Раньше диагноз ставили по запаху ацетона изо рта больного при гипергликемической коме ("запах гнилых яблок").