

Тема 10. Нуклеофильное замещение в ароматическом ядре
( к работам «-Нафтол», «2,4-Динитрофенилгидразин»)
Программа
Механизм SN2Ar и его общие закономерности. Механизм присоединения-элиминироавния (SN2АЕ) и элиминирования-присоединения (SN2ЕА). Дегидробензол: электронное строение, доказательства образования, включая перехват с помощью ловушек. Механизм SN1Ar. Нуклеофильное замещение гидрид-иона. Реакция Чичибабина. Акцепторы гидрид-иона.
Методические указания
Реакции этого типа могут быть представлены следующей общей схемой:
Ar-X + Nu- Ar-Nu + X-
Чаще всего замене подвергаются такие группы как Hal, SO3H, SO2Me, NO2 или диазониевая группа -NN+, отщепляющаяся в виде молекулы азота. Наиболее активными и распространенными нуклеофилами, как и в реакциях алифатических субстратов, являются ОН-, AlkO-, RNH2, RNHNH2, NH2-, OH-, RS- и т.п.
Различают три основных механизма нуклеофильного замещения в ароматическом ряду. Первые два в обобщенном виде обозначаются как SN2Ar, где приставка Ar отражает их специфику по сравнению с реакциями SN2 у насыщенного атома углерода. В свою очередь оба этих бимолекулярных механизма также имеют принципиальные отличия друг от друга. Один из них представляет собой процесс присоединения – элиминирования (SN2АЕ), тогда как другой – элиминирования – присоединения (SN2ЕА). Третий механизм относится к процессам мономолекулярного замещения и обозначается как SN1Ar. Существует также ряд других, менее распространенных механизмов, сведения о которых можно почерпнуть из цитированной ниже литературы.
Механизм присоединения–элиминирования (SN2АЕ). Это наиболее распространенный механизм и тип реакций. Главное условие его реализации - наличие в молекуле активирующих групп-электроноакцепторов, таких как NO2, NO, SO2Me, CN и т.п. На первой стадии происходит присоединение нуклеофила к субстрату 1 с образованием анионного -комплекса 2. Имеются многочисленные примеры фиксации -комплексов с помощью физических методов, главным образом ЯМР и УФ спектроскопии, в отдельных случаях их удалось выделить в кристаллическом состоянии и детально исследовать.
На второй стадии от -комплекса отщепляется уходящая группа X с парой электронов, в результате чего образуется продукт замещения 3. Поскольку первая стадия связана с разрушением стабильной ароматической системы, а вторая представляет собой энергетически выгодный процесс ароматизации, именно первая стадия определяет скорость всей реакции (k2>>k1). Реакция имеет второй суммарный порядок (первый по каждому реагенту) и скорость ее отвечает уравнению: v=k[ArX][Nu-].

Роль группы-активатора в субстрате двояка. С одной стороны, она увеличивает частичный положительный заряд на кольцевом атоме углерода, у которого происходит замещение, с другой – она стабилизирует -комплекс за счет дополнительной делокализации отрицательного заряда, привносимого нуклеофилом (вклад резонансных структур типа 2г весьма значителен). Действие групп-активаторов особенно ощутимо, когда они расположены в орто- и пара-положениях к реакционному центру. При наличии нескольких таких групп скорость нуклеофильного замещения сильно возрастает. Например, подвижность хлора в 2,4,6-тринитрохлорбензоле (пикрилхлориде) настолько велика, что он быстро превращается в пикриновую кислоту при не слишком осторожном хранении, т.е. реагирует с влагой воздуха.
Относительная подвижность галогенов в SN2-реакциях в алифатическом и ароматическом рядах резко различна. При замещении у насыщенного атома углерода галоген отрывается на скорость-определяющей стадии, поэтому легкость его отрыва определяется прочностью связи С-Hal: I>Br>Cl>F. В АЕ-реакциях в ароматическом ряду элиминирование происходит на второй, более быстрой стадии. Здесь прочность связи С-Hal уже не имеет решающего значения, и все определяется влиянием галогена на стадию присоединения нуклеофила. Есть три главных параметра, облегчающих эту стадию: увеличение положительного заряда на атоме, у которого протекает замещение, степень стабилизации -комплекса и стерический фактор (относительный размер входящей и уходящей групп). Ясно, что с этой точки зрения больше всего реакции должен благоприятствовать небольшой по размерам и наиболее электроотрицательный атом фтора. И действительно, относительные скорости замещения галогена при действии на 4-нитрогалогенбензолы метоксид-аниона при 50 оС равны F (312) >> Cl (1) > Br (0.74) > I (0.36). Та же последовательность характерна для других подобных реакций.
Механизм элиминирования-присоединения (SN2EA). Этот механизм реализуется в тех случаях, когда субстрат не содержит активирующих групп. Кроме того, нуклеофил должен быть сильным основанием. Чаще всего в качестве его выступает амид-ион, NH2- (pKa = 34) или металлорганические соединения. Например, хлор в хлорбензоле не обменивается на аминогруппу даже при длительном нагревании с аммиаком при температуре выше 200 оС. Однако при действии амида калия в жидком аммиаке такой обмен имеет место, хотя температура смеси не превышает –33 оС (температура кипения аммиака). Реакция имеет первый порядок по хлорбензолу и амид-иону и состоит из двух стадий, первая из которых в свою очередь включает две подстадии. Вначале амид-ион отщепляет один из орто-протонов, в результате чего образуется карбанион 4 (очевидно из-за близости галогена орто-связи С-Н более кислые, чем мета- и пара). Затем происходит элиминирование аниона Cl- и образуется нейтральная частица С6Н4 – бензол без двух атомов водорода. Она называется дегидробензолом или бензином (benzyne). Формально дегидробензол можно представить в виде биполярной структуры 5а. Однако фактически между свободными валентностями соседних орто-положений образуется нечто напоминающее ацетиленовую связь (структура 5б). Так как тройная связь внутри шестичленного цикла крайне напряжена, дегидробензол – очень неустойчив (его удалось заморозить и исследовать физическими методами лишь в аргоновой матрице при 8 оК). Поэтому он быстро присоединяет молекулу аммиака, в результате чего и образуется анилин. Вследствие симметричности тройной связи амид-ион с равной вероятностью присоединяется к любому из двух атомов углерода. Это было продемонстрировано на хлорбензоле, содержащем в положении 1 метку в виде углерода 14С (обозначен на схеме жирной точкой): в соотношении 1:1 были получены оба возможных анилина 6а и 6б, что и стало убедительным подтверждением правильности данного механизма. Из сказанного становится очевидным, что лимитирующей стадией при нуклеофильном замещении по механизму ЕА является стадия образования дегидробензола. А поскольку в ней участвуют субстрат и нуклеофил, реакция имеет второй суммарный порядок.

Замещение может протекать и по смешанному механизму. Например, на том же меченом хлорбензоле было показано, что при его нагревании с водным NaOH при 340 оС, 1-14С- и 2-14С-фенолы образуются в соотношении 58 и 42 %, соответственно. Из этого следует, что удельный вес механизмов ЕА и АЕ составляет 84 и 16 %.
Большинство производных дегидробензола (вместе с другими дегидроаренами их обобщенно называют аринами, соответственно ЕА-механизм иногда называют ариновым) имеют несимметричную ацетиленовую связь. Поэтому присоединение к ней нуклеофила приводит к различным продуктам, один из которых является перегруппировочным. Их соотношение может варьироваться в широких пределах в зависимости от электронной природы заместителя, например:

Влияние галогена в субстрате на легкость его замещения по ЕА-механизму носит сложный и неоднозначный характер. Так, при действии на галогенбензолы KNH2 в жидком аммиаке скорость реакции изменяется в ряду: Br>I>Cl>>F. В то же время, при действии металлорганических реагентов в апротонной среде эта последовательность почти полностью обращается F>Cl>Br>I. Очевидно в последнем случае лимитирующей является стадия депротонирования субстрата, поскольку его СН-кислотность будет тем выше, чем больше электроотрицательность галогена. Что касается первого случая, то относительная активность галогенов здесь отражает компромисс двух факторов: прочность связи С-Hal и –I-эффекта галогена.
Механизм SN1Ar. Этот механизм достоверно установлен лишь для реакции гидролиза солей арилдиазония, приводящей к образованию фенолов. Тем не менее, ввиду важности его алифатического аналога, ему придается фундаментальное значение и в ароматическом ряду. Процесс состоит из двух стадий: стадии генерирования арильного карбокатиона в результате относительно медленного гетеролиза связи Ar-N2+ и стадии последующего быстрого взаимодействия карбокатиона с водой.
![]()
Очевидно, концентрация нуклеофила никак не влияет на скорость всей реакции, которая поэтому описывается уравнением v = k[Ar-N2+]. Несомненно, основной причиной такого развития событий является стабильность молекулы азота, отщепляющейся на первой стадии. С другой стороны, причины, по которым этот механизм не реализуется в случае других ароматических субстратов, например, арилгалогенидов, заключаются в недостаточной устойчивости арильных карбокатионов (их свободная орбиталь относится к -типу и не стабилизируется сопряжением) и прочности связи Ar-X вследствие сопряжения заместителя X с кольцевой -системой.
Нуклеофильное замещение гидрид-иона. В отличие от реакций электрофильного замещения, где обычно вытесняется атом водорода, нуклеофильное замещение водорода протекает более трудно и специфично. Это обусловлено тем, что протон является хорошей уходящей группой, тогда как гидрид-ион, будучи сильным основанием (pKa 32), - одна из самых плохих уходящих групп. Тем не менее, известно немало реакций данного типа. Некоторые из них имеют важное синтетическое значение.
Как и в случае других групп, замещению гидрид-иона способствуют присутствующие в субстрате электроноакцепторные заместители. Особенно распространены такие реакции в азаароматических гетероциклах, например, пиридине 7, хинолине 9, пиримидине 10 или 1,3,5-триазине 11. Дело в том, что двоесвязанный внутрициклический атом азота (его называют также пиридиновым гетероатомом или просто азагруппой) является хорошим электроноакцептором, по силе сопоставимым с группой NO2. Однако в отличие от последней он не так подвержен стерическим эффектам, разнообразным процессам гидрирования и, что особенно важно, при протонировании может легко и обратимо давать катион. Например, катион пиридиния 8, способен реагировать с нуклеофилами на несколько порядков быстрее нейтральной молекулы.

Так, нитробензол при действии амида натрия образует сложную смесь смол и продуктов восстановления нитрогруппы. Содержание в ней о- и п-нитроанилинов – желаемых продуктов нуклеофильного замещения – незначительно. В то же время, пиридин при нагревании с амидом натрия с выходом более 70 % дает 2-аминопиридин 12. Эта реакция, открытая А.Е. Чичибабиным, - классический пример эффективного нуклеофильного замещения гидрид-иона.

Почти все реакции, где замещается гидрид-ион, начинаются с образования -комплекса и в этом отношении они мало чем отличаются от других реакций SN2AE. Их главная специфика приходится на заключительную стадию, где происходит элиминирование гидрид-иона. Различают три разновидности этого процесса: 1) термическое отщепление, 2) окислительное и 3) автоароматизацию. Термическое (самопроизвольное) элиминирование требует высокой температуры и встречается главным образом у аддуктов ароматических субстратов с металлорганическими соединениями. Последнее вполне вероятно, поскольку нуклеофил в этом случае должен быть еще более сильным основанием, чем гидрид ион. Например, аддукты пиридина с литийалкилами при длительном нагревании в апротонной среде отщепляют гидрид лития и превращаются в 2-алкилпиридины. По-видимому, аналогичным образом бензол с выходом 15 % превращается в трет-бутилбензол при нагревании в трет-бутиллитием:
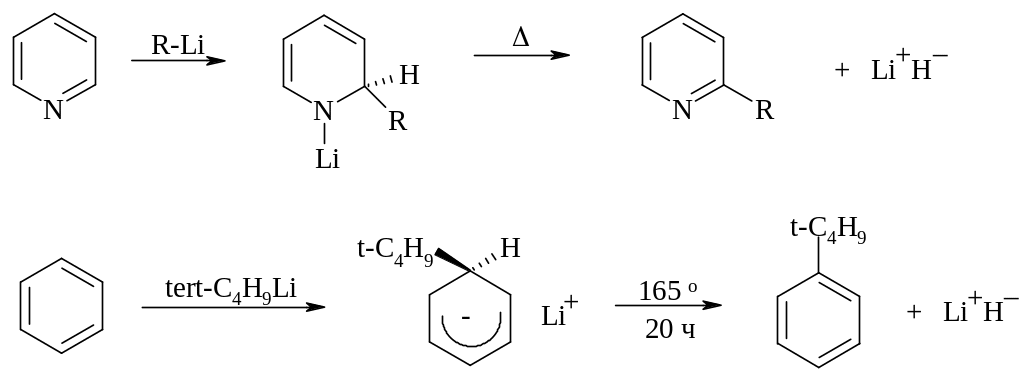
Значительно более широкий круг нуклеофилов можно использовать, если в реакционной смеси присутствуют различные акцепторы гидрид-иона. Ими могут быть специально добавляемые окислители: KMnO4, K3Fe(CN)6, хиноны, кислород и т.п. Как правило, реакция в этом случае протекает в мягких условиях и с хорошим выходом. Так, при действии на нитробензол едкого кали и воды в жидком аммиаке с последующим пропусканием кислорода образуется смесь орто- и пара-нитрофенолов в соотношении 3:1 и с суммарным выходом 66 %.
 Другой
пример – окислительное аминирование
пиримидинов амидом калия в жидком
аммиаке в присутствии перманганата
калия. Последний метод аминирования
электронодефицитных гетероциклов
получил в последние годы особенно
широкое применение.
Другой
пример – окислительное аминирование
пиримидинов амидом калия в жидком
аммиаке в присутствии перманганата
калия. Последний метод аминирования
электронодефицитных гетероциклов
получил в последние годы особенно
широкое применение.

Акцепторами гидрид-иона могут быть и вспомогательные группы, специально вводимые в субстрат или привносимые реагентом. Например, давно известна реакция аминирования нитроаренов гидроксиламином. Она протекает в присутствии щелочи, которая, по-видимому, способствует превращению гидроксиламина в более активный нуклеофил HONH-. Полагают, что при ароматизации -комплекса 13 в соответствии с приведенной ниже схемой отщепляется молекула воды. При этом, хотя формально в конечном итоге замещается гидрид-ион, более вероятно, что отщепляющимися частицами фактически являются протон и анион ОН-.

Сходная ситуация имеет место и в случае реакции Чичибабина. В анионном -комплексе 14 акцептором гидрид иона становится аминогруппа, обладающая слабой, но все же достаточной для взаимодействия с гидрид ионом NH-кислотностью. В результате происходит весьма редкое для органических реакций явление – выделение молекулярного водорода. Непосредственным продуктом аминирования является натриевая соль амина 15, которая после разложения водой через неустойчивый имино-таутомер 16 превращается в амин 12.

Чрезвычайно легко вступают в реакции нуклеофильного замещения водорода соли N-метоксипиридиния 17. Их можно легко получить метилированием доступного пиридин-N-оксида. При действии на них самых разнообразных нуклеофилов образуется аддукт типа 18, который моментально ароматизуется с отщеплением молекулы метанола. Трудно сказать отщепляется ли здесь водород в виде протона или гидрид-иона, но в любом случае процесс формально представляет собой замещение иона Н-. Известно много других разновидностей данной реакции.

Рекомендуемая литература
Дж. Марч. // Органическая химия.- М.: Мир, 1987,.- Т. 3.- С. 5-53.
Дж. Робертс, М. Кассерио // Основы органической химии.- М.: Мир, 1978.- Т. 2.- С. 244-254.
Ф. Кери, Р. Сандберг // Углубленный курс органической химии.- М.: Мир, 1981.- Кн. 2.- С. 241-251.
А.Ф. Пожарский, А.М. Симонов // Аминирование гетероциклов по Чичибабину.- Ростов-на-Дону: Изд-во РГУ, 1971.
А.Ф. Пожарский, А.М. Симонов, В.Н. Доронькин // Успехи химии.- 1978.- Т. 47.- № 11.- С. 1933-1969.
А.Ф. Пожарский // Теоретические основы химии гетероциклов.- М.: Химия, 1985.- C.
O.N. Chupakhin, V.N. Charushin, H.C. van der Plas // Nucleophilic Aromatic Substitution of Hydrogen.- San-Diego: Academic Press, 1994.
Х. Ван дер Плас // Химия гетероцикл. соедин.- 1987.- № 8.- C. 1011-1027.
A.R. Katritzky, A.F. Pozharskii // Handbook of Heretocyclic Chemistry, 2nd edition.- Pergamon, 2000.
* Подробнее об использование переходных металлов в органическом синтезе см. методические указания к курсу органической химии А.Ф. Пожарского, А.В. Гулевской «Новые реакции образования углерод-углеродных связей, катализируемые переходными металлами», Ростов-на-Дону, 2000 г.