

Министерство образования Российской Федерации
Ростовский государственный университет
Кафедра органической химии
А.Ф. Пожарский, о.В. Дябло
Методические указания
к практикуму по органической химии
часть вторая
(издание 2-е)
Ростов-на-Дону, 2001
Печатается по решению кафедры органической химии Ростовского государственного университета, протокол № 4-01/02 от 8 октября 2001 г.
Содержание
Тема 7. Электрофильное замещение в ароматическом ядре…………. 4
Тема 8. Фенолы………………………………………………………….. 13
Тема 9. Соли диазония ………………………………………………….. 24
Тема 10. Нуклеофильное замещение в ароматическом ядре…………. 32
Тема 7. ЭЛЕКТРОФИЛЬНОЕ ЗАМЕЩЕНИЕ В АРОМАТИЧЕСКОМ ЯДРЕ
(к синтезам «п-Нитроацетанилид», «п-Броманизол»)
Программа
Причины склонности ароматических соединений к реакциям электрофильного замещения. Типы электрофилов. Роль катализаторов (кислот Льюиса) при электрофильном замещении. -Комплексы и факторы, определяющие их устойчивость. Реакции нитрования, галоидирования, азосочетания, алкилирования и ацилирования по Фриделю-Крафтсу. Правила ориентации в моно- и дизамещенных бензола. Группы орто- пара- и мета-ориентанты. Активация и дезактивация бензольного кольца по отношению к электрофилам. Термодинамический и кинетический контроль в некоторых реакциях электрофильного замещения и его причины. -Комплексы и катион-радикалы в реакциях электрофильного замещения.
Методические указания
Общие положения. Реакции электрофильного замещения – превращения вида:
ArH + E+ ArE + H+
где ArH – молекула субстрата, E+ - электрофильный реагент. Большинство реакций электрофильного замещения требует применения катализатора – обычно кислоты Льюиса. Роль катализатора состоит в активации электрофильного агента путем создания или увеличения на нем положительного заряда. Например, соли железа в реакции бромирования способствуют поляризации связи Br-Br:
![]()
Аналогично, кислоты при алкилировании бензола олефинами протонируют последние, что ведет к образованию активного электрофила – карбокатиона.
![]()
Реакции электрофильного замещения включают две главные стадии – присоединение электрофила к бензольному кольцу с образованием -комплекса (об образовании -комплексов см. ниже) и стадию отщепления протона (ароматизацию), ведущую к конечному продукту:

Для большинства реакций электрофильного замещения скорость-определяющей является первая стадия. Кинетическое уравнение реакции имеет вид v = k [ArH][E+], т.е. она второго суммарного порядка, первого по субстрату и электрофилу. Поэтому реакции электрофильного замещения в ароматическом ряду обозначают символом SE2Ar.
-Комплекс – это фактически резонансно-стабилизированный карбениевый ион. В нем связь С-Е осуществляется за счет пары -электронов, ранее принадлежащих бензольному кольцу. Четыре оставшихся -электрона делокализованы на пяти углеродных атомах. Строение -комплекса может быть представлено как мезомерной структурой, показанной выше на схеме, так и набором следующих резонансных структур:

Правила ориентации в монозамещенных бензола. В монозамещенных бензола заместитель ориентирует вступающий электрофил преимущественно в орто-пара или мета-положения. Соответственно все заместители подразделяются на два больших класса: орто-пара-ориентанты и мета-ориентанты. орто-пара-Ориентирующей способностью обладают группы, имеющие доступные -электроны или неподеленные электронные пары, которые они могут предоставлять в сопряжение с бензольным кольцом. Это группы - -доноры. К ним, в частности, относятся:
ОН, ОR, NH2, NHR, NR2, F, Cl, Br, I, CH2=CH, CHC, C6H5
В результате донирования этими группами электронов в орто- и пара-положениях бензольного кольца наводится отрицательный -заряд. Поэтому именно сюда направляется атака электрофила.

Особняком стоят метил и другие алкильные группы, у которых нет -электронов или неподеленных электронных пар, но которые также являются орто-пара-ориентантами. Алкильные группы слабые индуктивные доноры. Кроме того, -электроны связей С-Н, смежных с бензольным кольцом, могут, хотя и слабо, участвовать в сопряжении с -электронами кольца. Это так называемое ,-сопряжение, которое полезно сравнить с ,-сопряжением:

Поскольку орто-пара-ориентанты увеличивают -электронную плотность в ядре, они активируют последнее к электрофильному замещению по сравнению с бензолом. Исключение составляют галогенбензолы, которые реагируют с электрофилами труднее бензола. Это объясняется тем, что в них –I-эффект галогена превалирует над +М-эффектом, т.е. галогены – более сильные -акцепторы, чем -доноры. Такие группы как ОН, OR, NH2, NR2 тоже обладают –I-эффектом, но он у них слабее +М-эффекта. В результате эти группы активируют бензольное кольцо:

По способности активировать бензольное кольцо к электрофильному замещению орто-пара-ориентанты образуют следующий ряд:
O- > NH2 > OH > OCH3 > NHCOCH3 > CH3 > C6H5, CH2=CH > Hal
Все мета-ориентанты являются электроноакцепторами. Они делятся на две категории:
группы с кратными связями, обладающие –I и –М-эффектом (так называемые электромерные заместители):
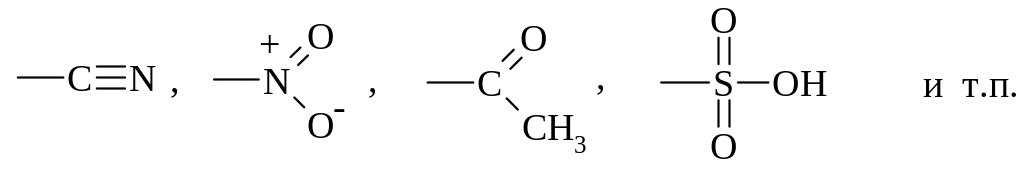
группы, не содержащие кратных связей и обладающие только –I-эффектом (индуктивные заместители):
![]()
Схема -электронного взаимодействия электромерных заместителей и бензольного кольца:

Таким образом, положительный -заряд наводится в орто- и пара-положениях, в результате чего электрофил атакует мета-положения, менее подверженные влиянию электроноакцепторной группы. По сравнению с бензолом мета-ориентанты дезактивируют кольцо к электрофильному замещению. Поэтому при наличии в кольце группы-дезактиватора многие реакции электрофильного замещения (алкилирование и ацилирование по Фриделю-Крафтсу, формилирование по Вильсмейеру, азосочетание и др.) становятся невозможными. По дезактивирующей способности мета-ориентанты располагаются в такой последовательности:
+
N(CH3)3 > NO2 > SO2CH3 > CN > CF3 > COCH3 > COOH > CHO
Правила ориентации в дизамещенных бензола. В дизамещенных бензола место вступления заместителей зависит от природы заместителей, силы их влияния и взаимного расположения. Наиболее простыми являются случаи так называемой согласованной ориентации, когда два заместителя ориентируют электрофил в одно и то же положение:

Во всех других случаях ориентация носит несогласованный характер (пунктирные стрелки в приведенных ниже структурах показывают менее предпочтительные места вступления электрофила):

Место вступления нового заместителя в дизамещенных бензола можно определить исходя из следующих правил:
Согласованное влияние оказывают заместители одного типа, когда они находятся в мета-положении или заместители разных типов, если они находятся в орто-пара-положениях друг к другу.
Несогласованная ориентация имеет место, когда однотипные заместители расположены в орто-пара-положениях друг к другу, а заместители различных типов – в мета-положении.
В случае несогласованной ориентации заместителей разных типов место вступления электрофила диктует орто-пара-ориентант.
При несогласованной ориентации заместителей одного типа место вступления электрофила определяет более сильный ориентант.
Из-за пространственных помех электрофил практически не вступает в положения, расположенные между двумя заместителями. Таковы, например, положения 2 в резорцине и в мета-нитроацетанилиде.
Выше явления ориентации рассматривались с точки зрения распределения зарядов в бензольном кольце. Есть альтернативный и, строго говоря, более последовательный способ анализа, основанный на рассмотрении факторов, способствующих стабилизации или дестабилизации соответствующих -комплексов. Так, при электрофильном орто-пара-замещении в катионе триметиланилиния образующийся -комплекс неустойчив, поскольку положительный заряд в -системе делокализован таким образом, что часть его находится на атоме, с которым связан положительно заряженный азот (см. правую из трех представленных ниже резонансных структур):
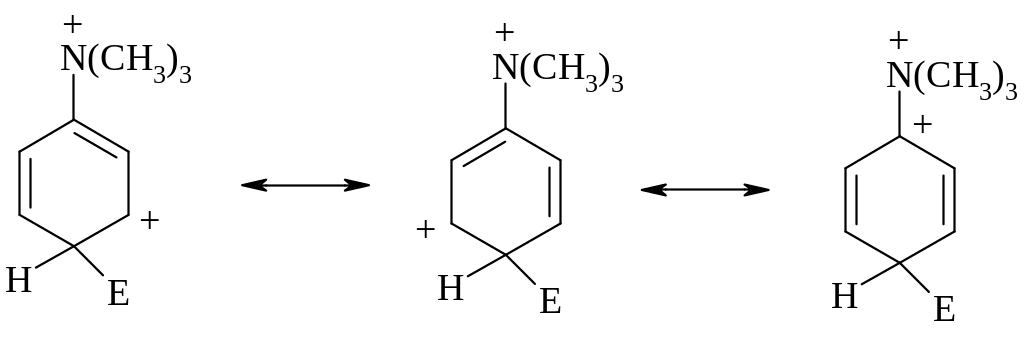
С другой стороны, при электрофильном замещении в мета-положение получается более устойчивый -комплекс, в котором положительные заряды на азоте и в -системе максимально разделены:

В результате энергетически более выгодным оказывается путь, когда электрофил присоединяется к мета-положению. Студентам следует разобрать аналогичные ситуации для электрофильного замещения в соединениях типа анилина, стирола, нитробензола. Рекомендуется также в каждом случае построить кривые потенциальной энергии, соответствующие замещению в различные положения.
Термодинамический и кинетический контроль в реакциях электрофильного замещения. Как правило, в реакциях SE2Ar обратимой является только первая стадия. Однако в некоторых случаях (сульфирование, ацилирование) обратимость характерна и для второй стадии. Это приводит к последствиям двоякого рода. Во-первых, можно удалить уже введенный заместитель, что иногда бывает желательно с синтетической точки зрения. Этот прием, например, используют при получении 2,6-динитротолуола:

Как вы представляете себе механизм элиминирования сульфогруппы при кипячении 2,6-динитротолуол-4-сульфокислоты в воде?
Во-вторых, в реакциях с обратимой стадией ароматизации строение образующихся продуктов нередко зависит от температуры. Обычно при пониженной температуре реакция идет преимущественно в сторону более быстро образующегося изомера. Например, при сульфировании толуола при температуре ниже 100 оС больше образуется о-толуолсульфокислоты, чем п-толуолсульфокислоты. Это объясняется тем, что в толуоле отрицательный заряд несколько выше в орто-положениях, скорость сульфирования которых поэтому больше по сравнению с пара-положением. Кроме того, здесь работает так называемый статистический фактор: орто-положений два, а пара – одно, поэтому вероятность вхождения электрофила в орто-положение в отсутствии других факторов в принципе вдвое выше. Однако если проводить сульфирование при 120-150 оС практически единственным продуктом реакции оказывается п-толуолсульфокислота. Последняя в этих условиях термодинамически более устойчива и поэтому из-за обратимости второй стадии равновесие скатывается в ее сторону. Принято говорить, что при пониженной температуре процесс протекает в условиях кинетического контроля, а при повышенной – в условиях термодинамического контроля:

-Комплексы и катион-радикалы в реакциях электрофильного замещения. Приведенный выше механизм реакций электрофильного замещения несколько упрощен. В действительности, прежде чем образуется -комплекс между субстратом и электрофилом происходит более слабое взаимодействие, ведущее к образованию так называемого -комплекса (или «комплекса соударения»). В нем осуществляется слабое донорно-акцепторное взаимодействие между ВЗМО ароматической молекулы и НСМО электрофила. В таком комплексе электрофил как бы «зависает» над бензольным кольцом.
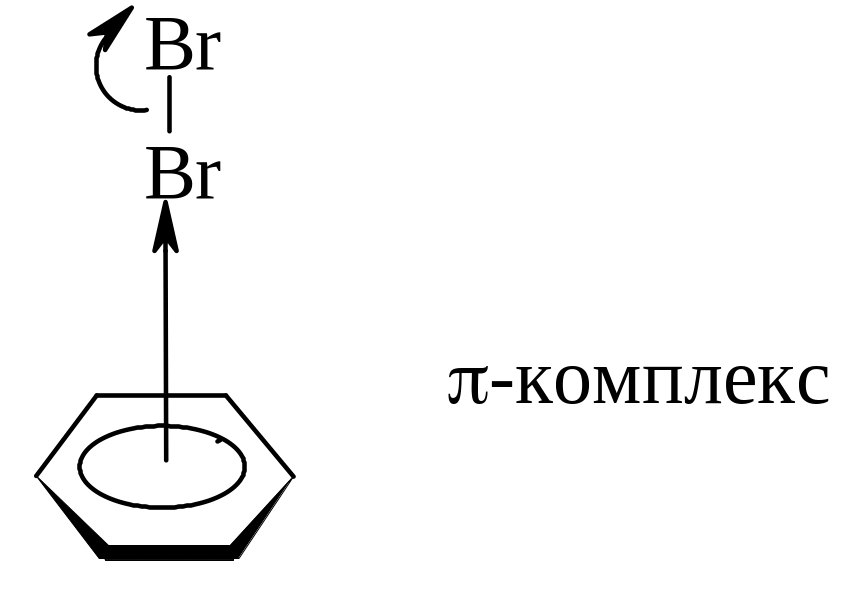
Энергия -взаимодействия донора и акцептора не превышает 3-5 ккал/моль, поэтому образование -комплекса обычно не вносит существенного вклада в общий энергетический баланс реакции (рис. 1). Как видно, переходное состояние (ПС1), отвечающее образованию -комплекса-I лежит намного ниже переходного состояния (ПС2), отвечающего образованию -комплекса. Предполагают, что отщепляющийся от -комплекса протон участвует в образовании -комплекса-II с продуктом замещения.

Рис. 1. Кривая потенциальной энергии реакции электрофильного замещения.
Однако иногда встречаются ситуации, когда -взаимодействие между субстратом и электрофилом весьма велико и в отдельных случаях даже приводит к переносу электрона с верхней занятой -орбитали субстрата на нижнюю свободную орбиталь акцептора. При этом субстрат переходит в катион-радикал, а электрофил в радикал (если первоначально он был катионом) или в анион-радикал (если исходный электрофил – нейтральная молекула). В этом случае вся реакция протекает, как правило, очень быстро и часто приводит к образованию в качестве побочного вещества димера исходной молекулы. Само по себе выделение таких димеров служит признаком реализации катион-радикального механизма электрофильного замещения, который в самом общем виде можно представить так:
1) ArH + E+ ArH + . + E.
2) ArH+ . + E. ArE + H+
3) ArH+ . ½ Ar-Ar + H+
Этот механизм характерен главным образом для ароматических субстратов содержащих сильные электронодонорные группы (например, N(CH3)2) и имеющих протяженную -систему. Типичный пример такой реакции – нитрование 1,8-бис(диметиламино)нафталина 1 («протонной губки») смесью HNO3–H2SO4. Образующийся на первой стадии катион-радикал «протонной губки» 2 можно представить набором нескольких резонансных структур, часть из которых (2 а-в) показана ниже на схеме. Как видно, спиновая плотность в катион-радикале рассредоточена по орто- и пара-положениям бензольных колец, причем в пара-положении, как показывают расчеты она больше. На второй стадии происходит рекомбинация катион-радикала 2 с радикалом NO2., причем реакция протекает главным образом по положению 4 нафталинового кольца с образованием нитропроизводного 3. Некоторая часть катион-радикалов успевает димеризоваться также по положению 4, давая бинафтил 4. В силу меньшей спиновой плотности в орто-положении, а также стерических факторов продукт замещения или димер по орто-положению нафталинового кольца не зафиксирован.


При димеризации катион-радикала 2 вначале образуется дигидропроизводное 5, которое быстро теряет два протона и превращается в соединение 4.

Хотя подавляющее большинство реакций SE2Ar сопровождается замещением атома водорода (отщепление в виде протона) известны и так называемые реакции ипсо-замещения, в которых отщепляющаяся группа – более тяжелый заместитель. Ипсо-замещению особенно подвержены такие группы как SO3H, COCH3, CO2H и некоторые другие (см стр.11 и 34, где даны примеры SE2Ar замещения SO3H , SCH3 и CH2OH групп).
Рекомендуемая литература
А.Н. Несмеянов, Н.А. Несмеянов // Начала органической химии.- М.: Химия, 1970.- Т. 2.- С. 31-48.
А. Терней // Современная органическая химия.- М.: Мир, 1981.- Т. 1.- С. 591-625.
Ф. Кери, Р. Сандберг // Углубленный курс органической химии- М.: Химия, 1981.- Кн.1.- С. 324-380.
Л.С. Эфрос, И.Я. Квитко //Химия и технология ароматических соединений в задачах и упражнениях.- М.: Химия, 1984.- С. 33-54.
М.В. Горелик, Л.С. Эфрос // Основы химии и технологии ароматических соединений.- М.: Химия, 1992.- С. 132-294.