

6.4 Основные теории гетерогенного катализа
Рассмотрим теперь, в чем заключается механизм каталитического действия. Первое истолкование катализа как процесса образования и разложения нестабильных соединений на поверхности дал еще в конце 19 века Сабатье (1897). Однако общей теории, объясняющей все аспекты катализа и позволяющей предсказать тип катализатора для новой реакции, не существует и в настоящее время.
Упрощенно природу активной поверхности можно представить в виде двух моделей: 1) активна вся поверхность (Лэнгмюр 1922, Боресков 1957) и 2) катализ протекает на активных центрах поверхности (Тэйлор 1925). Согласно Тэйлору, такие центры включают различные кристаллические структуры и/или дефектные участки на поверхности. Таким образом, основное различие между указанными двумя концепциями катализа сводится к оценке роли коллективных или локальных взаимодействий в катализе.
Представления об активных центрах в настоящее время получили общее признание и хорошо подтверждаются экспериментальными данными об отравлении катализатора микропримесями, так называемых, каталитических ядов. Так, например, при катализе на металлах и твердых кислотах отравление катализатора наступает при наличии примесей сернистых соединений и азотистых оснований, соответствующих нескольким процентам от всех атомов на поверхности или 3-5 мкмоль/м2 (катализатора).
Эти представления были детально проработаны в рамках мультиплетной теории Баландина (1929). В теории каталитического действия Баландина был проведен совместный анализ геометрического и энергетического соответствия между активными центрами и фрагментами реагирующих молекул. Мультиплетная теория была подтверждена множеством экспериментов для различных реакций на катализаторах разных типов, выполненных как в лаб. Баландина, так и в других центрах (работы Ридиила, Бека и др.). Таким образом, в рамках мультиплетной теории активные центры рассматриваются как организованная группа частиц на поверхности, соответствующая молекулам реагентов, как по размеру, так и по энергии разрываемых связей.
Рассмотрим геометрический аспект теории. Было установлено, что для реакции дегидроциклизации парафинов с образованием ароматических углеводородов активные центры представляют собой секстеты, т.е. 6 атомов металлов VIII группы, расположенных как в правильном шестиугольнике. Поэтому в монокристалле эта реакция протекает только на грани [111], на которой имеются такие конфигурации, а на грани [110] не идет.
Наиболее распространены активные центры типа дублетов, на которых протекают реакции типа элиминирования и присоединения, например, превращения спиртов на оксидах металлов. В молекуле спирта длина С-С связи 1,54А больше длины связи С-ОН, 1,43А, поэтому с увеличением параметра решетки оксида МО до 1,6-1,7А преимущественно протекает процесс дегидрирования, а уменьшение параметра решетки способствует дегидратации.
Кроме геометрического соответствия, также необходимо энергетическое, которое рассмотрим на примере реакции АВ + СD AC + BD, протекающей через активированный комплекс, типа приведенного ниже:
●
А ----- С

● ● (Z) где Z - активные центры
B ------ D
●
Используя закон Гесса, можно получить выражения для энергии образования активированного комплекса из исходных веществ ε1 и продуктов ε2 реакции
Кобозев (1946) разработал теорию катализа для предельно разбавленных систем типа металл/носитель. В этой теории активные центры представляют собой ансамбли с определенным числом атомов металла на поверхности (n). Например, для синтеза аммиака активные центры включают три атома железа (n =3), а для гидрирования олефинов нужны ансамбли с n =2. Атомы металла случайно распределены по миграционным ячейкам на поверхности носителя.
Согласно теории ансамблей каталитическая активность металла зависит от двух параметров n и р, где р - это отношение площади ячейки миграции к удельной поверхности катализатора. Поскольку в этой теории не учитываются электронные взаимодействия и природа металла, она имеет весьма ограниченную область применения.
В соответствии с
электронной теорией Ф.Ф. Волькенштейна,
рассмотрим связь каталитической
активности с электронными свойствами
твердого катализатора – полупроводника.
Как известно, в полупроводниках имеются
3 зоны: валентная, зона проводимости и
разделяющая их запрещенная зона, в
которой находится уровень Ферми – т.е.
уровень (ε)
электрохимического потенциала
полупроводника .
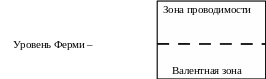
Увеличение концентрации электронов в полупроводник смещает уровень Ферми вверх, а увеличение концентрации дырок смещает уровень Ферми вниз, ближе к валентной зоне.
По теории
Волькенштейна k
= k0exp(-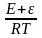 ),
где ε
- уровень Ферми. На положение ε
оказывают влияние как адсорбированные
на поверхности частицы, так и примеси,
введенные в твердый катализатор –
полупроводник, Ясно, что адсорбаты и
добавки доноры электронов будут смещать
уровень Ферми вверх, а адсорбаты и
добавки акцепторы электронов будут
смещать уровень Ферми вниз. Поэтому для
реакций с участием электронов и дырок
на лимитирующей стадии можно ожидать
различное влияние добавок (промоторов)
к катализатору. Действительно было
обнаружено, что добавка доноров
электронов оксидов алюминия или галлия
в катализатор ZnO
увеличивает проводимость ZnO
и скорость реакции H-D
обмена, протекающей на поверхности по
механизму:
),
где ε
- уровень Ферми. На положение ε
оказывают влияние как адсорбированные
на поверхности частицы, так и примеси,
введенные в твердый катализатор –
полупроводник, Ясно, что адсорбаты и
добавки доноры электронов будут смещать
уровень Ферми вверх, а адсорбаты и
добавки акцепторы электронов будут
смещать уровень Ферми вниз. Поэтому для
реакций с участием электронов и дырок
на лимитирующей стадии можно ожидать
различное влияние добавок (промоторов)
к катализатору. Действительно было
обнаружено, что добавка доноров
электронов оксидов алюминия или галлия
в катализатор ZnO
увеличивает проводимость ZnO
и скорость реакции H-D
обмена, протекающей на поверхности по
механизму:
H+ + D+ + 2e → H-D
В то же время введение добавок оксида щелочного металла, снижающая проводимость ZnO приводила к уменьшению активности катализатора.
Для реакции с участием дырок на лимитирующей стадии, напротив, введение добавок оксида щелочного металла в катализатор NiO увеличивает концентрацию дырок (p) и каталитическую активность в реакции разложения:
N2O → N2 + O- + p
2O- + 2p →O2
Однако введение добавок доноров электронов оксидов индия или галлия снижает активность NiO в этой реакции. Таким образом, для простых реакций на полупроводниках теория ФФВ позволяет управлять активностью катализаторов за счет введения добавок, влияющих на положение уровня Ферми.
Авторы радикальной теории катализа (Семенов, Воеводский) предлагали рассматривать поверхность катализатора как макрорадикал со свободными валентностями. Таким образом, катализатор возбуждает образование и развитие цепей, например, в реакциях высокотемпературного окисления и хлорирования углеводородов, действительно наблюдался выход радикалов в объем с поверхности катализатора:
RH + 2OZ → ROZ + HOZ
O2 + ROZ → RO2 + OZ
Представления о теории и эффектах катализа в конце ХХ века
Кластерные (геометрические) и лигандные (электронные) эффекты в катализе, сформулированные Sachtler (1973), по существу, представляют собой детализацию теории Баландина, причем их раздельное рассмотрение или противопоставление, по-видимому, не корректно.
Эффекты сильного взаимодействия в системе металл-носитель имеют значение для носителей, обладающих высокой реакционной способностью (например, оксиды Ti, Zr, V и др.), особенно при высоких температурах. Эти эффекты, главным образом, вызваны частичным восстановлением оксидного носителя с последующим сплавлением Ti, Zr, V (из носителя) с активным металлом катализатора. Однако для традиционных оксидных носителей (диоксид кремния, оксид алюминия и др.) при температуре ниже 850K этим эффектом можно пренебречь.
Классификация состава и структуры частиц на поверхности на примере полиметаллических катализаторов (Miura-1982). В зависимости от соотношения энергий взаимодействия между двумя металлами и их взаимодействия с носителем возможны следующие типы металл-содержащих частиц на поверхности носителя:
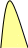

х+x +++ +++ ++ xx ++ xx

+x+x+ +x+x+ ++x++ +++xxx +++ xxx
+x+x+x +x+x+x+ ++xxx++ ++++xxxx ++++ xxxx
+x+x+x+x +xx+x+xx+ ++xxxxx++ +++++xxxxx +++++ xxxxx
I II III IV V
Рис. 1. Типы структур биметаллических катализаторов: I – гомогенная смесь металлов M1 (+) и M2 (x); II- сегрегация M1 поверхности частицы; III- экранирование поверхности M2 металлом M1 (“cherry model”); IV- контактная частица, содержащая 2 индивидуальных металла; V – отдельные частицы индивидуальных металлов
Структуры I-III образуются, когда энергия взаимодействия между двумя металлами выше их энергии взаимодействия с носителем. В противном случае образуются структуры IV и V.
Трансформации структур I-III могут быть вызваны различиями между M1 и M2, такими как теплота сублимации, дисперсность частиц и др. Однако наиболее сильной побудительной причиной таких трансформаций является воздействие среды, окружающей катализатор. Ясно, что в случае лабильных систем типа I-III состав поверхности может значительно изменяться при переходе от вакуума к восстановительной (Н2), окислительной (О2) или углеводородной среде. В свою очередь, каталитические свойства поверхности главным образом определяются составом поверхности. Таким образом, мы приходим к выводу о необходимости исследования изменений состава поверхности при взаимодействии с компонентами реакционной смеси. Эти реакции относятся к топохимическим и мы рассмотрим их позже.
В заключение обсуждения теорий катализа подчеркнем, что основной целью их создание является создание научно обоснованных подходов к разработке наиболее эффективных катализаторов для основных процессов нефтепереработки, нефтехимии, общей и органической химии.
Рассмотрим более подробно основные каталитические процессы нефтепереработки и нефтехимии. После перегонки нефти с отбором «светлых» дистиллятов, остатки (мазут и вакуумные дистилляты) направляют на установку каталитического крекинга (1). Фракции н.к. (35-85˚С) и нафты (85-180˚С) направляют на установки изомеризации (2) и каталитического риформинга (3). Кроме того, на современных нефтеперерабатывающих заводах имеются каталитические процессы гидрокрекинга (4) алкилирования (5) бутенов, а также синтез оксигенатов (6), т.е. МТБЭ, ДИПЭ и МТАЭ (метил-трет-амиловый эфир). Для получения продуктов соответствующего качества сырье каталитических процессов, а также товарные продукты подвергают гидроочистке (устраняя соединения серы, азота, коксогены и др.).
1. Каталитический крекинг представляет собой наиболее крупнотоннажный процесс нефтепереработки; во всем мире в этом процессе получают основной компонент высокооктанового бензина. В настоящее время каталитический крекинг осуществляют на цеолитсодержащих катализаторах типа НZSM при температуре 520-530˚С и объемной скорости подачи сырья в реактор 2-3 ч-1. Конверсия сырья обычно составляет 75-78%. Катализатор непрерывно регенерируют путем выжига кокса в регенераторе при 650-730˚С. Состав продуктов, об.%:
газы С2-С4 24-26
бензин 59-62 (ОЧ ис.м. до 95)
газойль 16-18
Лекция 16_08
6. .4. Легкие олефины из метанола (МТО)
Метанол является перспективным альтернативным (не нефтяным) сырьем для получения олефинов (цена метанола обычно ниже, чем нафты). На фирме Mobil были разработаны два варианта превращения метанола: двухстадийный в неподвижном слое катализатора (1) дегидратация до ДМЭ и 2) превращение ДМЭ в олефины) и одностадийный способ в кипящем слое, аналогично ФКК. В качестве катализатора используют модифицированный цеолит ZSM5. Полное превращение метанола реализуется при 500-600˚С, причем селективность по олефинам (C2-C5) превышает 80%.
СН3OH à[СН2 --Н2O]à aС2Н4 + bС3Н6 + dС4Н8 + (2a+3b+4d)Н2O
где отношение этилен/пропилен (a/b)~0,6-2,0 и выход бутенов доходит до 18% (a/d~1,2-8).
Важно, что в продуктах отсутствует бутадиен (при крекинге 4-9%); и, кроме того, образуется очень мало жидких продуктов – 3,5% (20-30%) и метана 0,5-4% (13-17%). Единственный недостаток процесса – это быстрая деактивации катализатора коксом. Характеристика селективности процесса МТО ниже сопоставлена с селективностью для крекинга СНГ с водяным паром (в скобках)
Легкие олефины из метанола (х >99%). Сопоставление процессов
Процесс Селективность |
MTO MOBIL |
UNION CARBIDE |
IFP |
по:Этилену Пропилену |
21 38.7 |
53 27.1 |
35.8 39.7 |
Бутены Олефины C5-C6 |
18.5 2.4 |
6.4 3.5 |
8.6 1.1 |
Этилен + Пропилен Сумма олефинов |
59.7 80.6 |
80.1 90 |
75.5 85.2 |
CО2 метан C2-C5 параф. C6+ не-арены Арены |
- 0.5 15.6 1.3 2.0 |
5.0 3.9 1.1 - - |
- 3.6 11.2 - - |
5. Прямое превращение метана в олефины
В разработке этого процесса принимают участие многие фирмы и лаборатории. Действительно, благодаря такому способу можно получать сырье для нефтехимии не из все более дефицитного нефтяного сырья, а из доступного природного газа. Процесс окислительной димеризации метана с получением этилена может быть описан следующим брутто уравнением:
2 CH4 + O2 à C2H4 + H2O + COx
Обычно этот процесс протекает в присутствии оксидов щелочных и щёлочноземельных металлов при довольно высокой температуре (850-900˚С) и значительных временах контакта. Недостатки процесса низкая производительность (менее 0,1 ч-1) и селективность, максимальный выход олефинов не превышает 20%. Гораздо более перспективным процессом является непрямое превращение природного газа в олефины через метанол.
Таким образом, современная нефтехимия располагает рядом хороших процессов, обеспечивающих получение олефинов из различных видов сырья - нафты, СНГ и природного газа (через метанол). Теперь могут быть использованы рассмотрим основные направления превращений олефинов в различные продукты нефтехимии.