

,Характеристика свойств элемента и его соединений по электронной формуле и по положению в периодической системе элементов.
Зная формулировка периодического закона и используя периодическую систему элементов Д. И. Менделеева, можно дать характеристику любому химическому элементу и его соединениям. Такую характеристику химического элемента удобно складывать по плану. I. Символ химического элемента и его название. II. Положение химического элемента в периодической системе элементов Д.И. Менделеева: 1) порядковый номер; 2) номер периода; 3) номер группы; 4) подгруппа (главная или побочная). III. Строение атома химического элемента: 1) заряд ядра атома; 2) относительная атомная масса химического элемента; 3) число протонов; 4) число электронов; 5) число нейтронов; 6) число электронных уровней в атоме. IV. Электронная и электронно-графическая формулы атома, его валентные электроны. V. Тип химического элемента (металл или неметалл, s-, p-, d-или f-элемент). VI. Формулы высшего оксида и гидроксида химического элемента, характеристика их свойств (основные, кислотные или амфотерные). VII. Сравнение металлических или неметаллических свойств химического элемента со свойствами элементов-соседей по периоду и подгруппой. VIII. Максимальный и минимальный степень окисления атома. Например, предоставим характеристику химического элемента с порядковым номером 15 и его соединениям по положению в периодической системе элементов Д. И. Менделеева и строению атома. I. Находим в таблице Д. И. Менделеева клетку с номером химического элемента, записываем его символ и название. Химический элемент номер 15 - Фосфор. Его символ Р. II. Охарактеризуем положение элемента в таблице Д. И. Менделеева (номер периода, группы, тип подгруппы). Фосфор находится в главной подгруппе V группы, в 3-м периоде. III. Предоставим общую характеристику состава атома химического элемента (заряд ядра, атомная масса, число протонов, нейтронов, электронов и электронных уровней). Заряд ядра атома фосфора равен +15. Относительная атомная масса фосфора равна 31. Ядро атома содержит 15 протонов и 16 нейтронов (31 - 15 = 16. Атом фосфора имеет три энергетических уровня, на которых находятся 15 электронов. IV. Составляем электронной и электронно-графическую формулы атома, отмечаем его валентные электроны. Электронная формула атома фосфора: 15P 1s2 2s2 2p6 3s2 3p3. Электронно-графическая формула внешнего уровня атома фосфора: на третьем энергетическом уровне на 3s-подуровня находятся два электрона (в одной клетке записываются две стрелки, имеющие противоположное направление), на три р-подуровне находятся три электрона (в каждой из трех клеток записываются по одной стрелке, имеющие одинаковое направление). Валентными электронами являются электроны внешнего уровня, т.е. 3s2 3p3 электроны. V. Определяем тип химического элемента (металл или неметалл, s-, p-, d-или f-элемент). Фосфор - неметалл. Поскольку в последнее подуровнем в атоме фосфора, который заполняется электронами, является p-подуровень, Фосфор относится к семейству p-элементов. VI. Составляем формулы высшего оксида и гидроксида фосфора и характеризуем их свойства (основные, кислотные или амфотерные). Высший оксид фосфора P2O5, проявляет свойства кислотного оксида. Гидроксид, соответствующий высшему оксиду, H3PO4, проявляет свойства кислоты. Подтвердим указанные свойства уравнениями видповиних химических реакций: P2O5 + 3 Na2O = 2Na3PO4 H3PO4 + 3NaOH = Na3PO4 + 3H2O VII. Сравним неметаллические свойства фосфора со свойствами элементов-соседей по периоду и подгруппой. Соседом фосфора по подгруппе являются азот. Соседями фосфора за периодом является кремний и Сера. Неметаллические свойства атомов химических элементов главных подгрупп с ростом порядкового номера растут в периодах и снижаются в группах. Поэтому неметаллические свойства фосфора более выражены, чем у кремния и менее выражены, чем у азота и серы. VIII. Определяем максимальный и минимальный степень окисления атома фосфора. Максимальный положительный степень окисления для химических элементов главных подгрупп равен номеру группы. Фосфор находится в главной подгруппе пятой группы, поэтому максимальная степень окисления фосфора +5. Минимальная степень окисления для неметаллов в большинстве случаев равен разнице между номером группы и числом восемь. Так, минимальная степень окисления фосфора -3.
Ковалентная химическая связь. Механизмы её образования, характеристика ковалентной связи.
В слове "ковалентная" приставка "ко-" означает "совместное участие". А "валента" в переводе на русский – сила, способность. В данном случае имеется в виду способность атомов связываться с другими атомами.
При образовании ковалентной связи атомы объединяют свои электроны как бы в общую "копилку" – молекулярную орбиталь, которая формируется из атомных оболочек отдельных атомов. Эта новая оболочка содержит по возможности завершенное число электронов и заменяет атомам их собственные незавершенные атомные оболочки.
Рассмотрим возникновение ковалентной связи на примере образования молекулы водорода из двух атомов водорода (рис. 3-1). Этот процесс уже является типичной химической реакцией, потому что из одного вещества (атомарного водорода) образуется другое – молекулярный водород. Внешним признаком энергетической выгодности этого процесса является выделение большого количества теплоты.
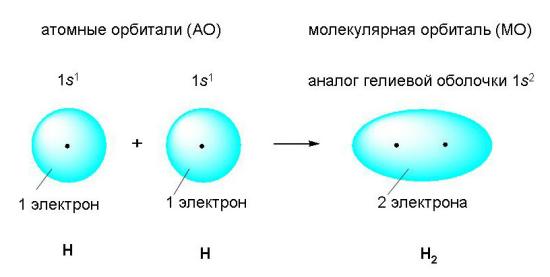
Рис 3-1. Возникновение ковалентной связи при образовании молекулы водорода из двух атомов водорода. Электронные оболочки атомов водорода (с одним s-электроном у каждого атома) сливаются в общее электронное облако (молекулярную орбиталь), где оба электрона "обслуживают" ядра независимо от того, "свое" это ядро или "чужое".
Когда электронные оболочки двух атомов водорода сближаются и образуют новую, теперь уже молекулярную электронную оболочку (рис. 3-1), эта новая оболочка подобна завершенной электронной оболочке атома благородного газа гелия (1s2).
Завершенные оболочки, как мы помним, устойчивее незавершенных. Таким образом, суммарная энергия новой системы – молекулы водорода – оказывается гораздо ниже суммарной энергии двух несвязанных атомов водорода. Избыток энергии при этом выделяется в виде теплоты.
Минимум энергии молекулы отвечает определенному расстоянию между ядрами атомов водорода (рис. 3-2). Если атомы в молекуле с помощью внешней силы сдвинуть еще ближе, то в действие вступает мощное отталкивание между одноименно заряженными ядрами атомов и общая энергия системы начинает быстро возрастать. Это невыгодно системе, поэтому длина связи представляет собой строго определенное, равновесное значение. Для молекулы водорода равновесная длина химической связи составляет 0,74 ангстрема (1 А = 10–8 см), как это видно на рис. 3-2.
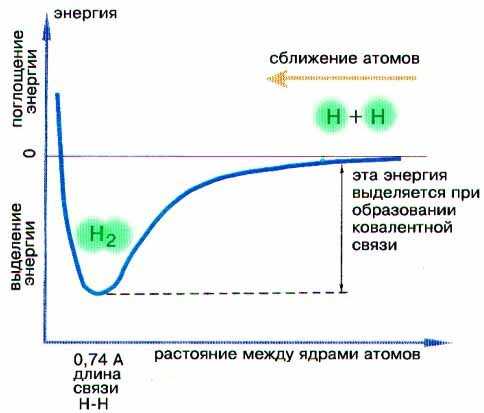
Рис. 3-2. Кривая изменения потенциальной энергии при взаимодействии двух атомов водорода с образованием молекулы водорода.
В образовавшейся системе из двух водородных атомов каждое ядро обслуживается двумя электронами. В новой (молекулярной) оболочке уже невозможно различить, какой из электронов ранее принадлежал тому или другому атому. Принято говорить, что электроны обобществлены.
Поскольку оба ядра претендуют на пару электронов в равной степени, электронная плотность сосредоточена как вокруг ядер, так и в пространстве между атомами (это показано на рис. 3-3). Именно эту область повышенной электронной плотности между ядрами и называют ковалентной связью.
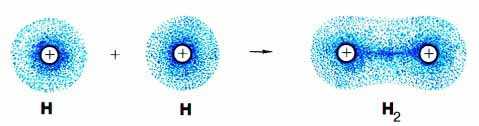
Рис. 3-3. Другой способ изображения атомных и молекулярной орбиталей: густота точек отражает "электронную плотность", то есть вероятность нахождения электрона в какой-либо точке пространства около ядер атомов водорода. Видно, что значительная электронная плотность сосредоточена в пространстве между двумя ядрами в молекуле водорода.
На рисунках 3-1 и 3-3 вы видите очень детальное изображение ковалентной связи. На практике используют более простые способы. Например, американский химик Дж. Льюис в 1916 году предложил обозначать электроны точками рядом с символами элементов. Одна точка обозначает один электрон. В этом случае образование молекулы водорода из атомов записывается так:
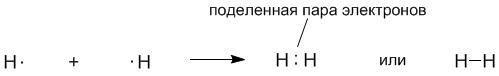
Оказалось, что формулы Льюиса имеют глубокий химический смысл. Мы видим, что связь между атомами водорода обозначается парой электронов. Как предположил Льюис, именно пара электронов позволяет образовать ковалентную связь. Впоследствии это предположение подтвердилось квантовой теорией.
Ковалентной связью называется связывание атомов с помощью общих (поделенных между ними) электронных пар.
Рассмотрим связывание двух атомов хлора 17Cl (заряд ядра Z = 17) в двухатомную молекулу с позиций строения электронных оболочек хлора. Для этого запишем формулу Льюиса для атома хлора и конфигурацию его внешней электронной оболочки:

На внешнем электронном уровне хлора содержится s2 + p5 = 7 электронов. Поскольку электроны нижних уровней не принимают участия в химическом взаимодействии, точками мы обозначили только электроны внешнего, третьего уровня. Эти внешние электроны (7 штук) можно расположить в виде трех электронных пар и одного неспаренного электрона.
После объединения атомов в молекулу из двух неспаренных электронов атомов получается новая электронная пара:
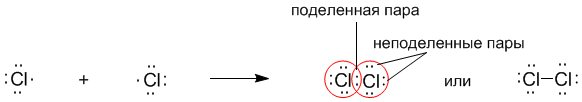
При этом каждый из атомов хлора оказывается в окружении ОКТЕТА электронов. В этом легко убедиться, если обвести кружком любой из атомов хлора.
Ковалентную связь образует только пара электронов, находящаяся между атомами. Она называется поделенной парой. Остальные пары электронов называют неподеленными парами. Они заполняют оболочки и не принимают участие в связывании.
Это правило применительно к химическим связям можно сформулировать так:
атомы образуют химические связи в результате обобществления такого количества электронов, чтобы приобрести электронную конфигурацию, подобную завершенной электронной конфигурации атомов благородных элементов.
Два атома водорода, объединившись в молекулу, приобрели “завершенную” молекулярную оболочку, подобную завершенной электронной оболочке атома благородного газа гелия (1s2). Атомы хлора в молекуле приобрели молекулярную оболочку, похожую на завершенную оболочку атома аргона (...3s23p6).
Познакомимся теперь с ионной связью. Как ни удивительно, она ничем принципиально не отличается от ковалентной связи. Движущей силой ее образования является все то же стремление атомов к октетной оболочке. Но в ряде случаев такая “октетная” оболочка может возникнуть только при передаче электронов от одного атома к другому. Поэтому ионная связь, в отличие от ковалентной, возникает только между атомами разного вида.
Рассмотрим конкретный пример: реакцию между атомами натрия (Z = 11) и фтора (Z = 9). При образовании связи между ними оба элемента приобретают внешнюю электронную оболочку благородного газа неона (Z = 10). Для того, чтобы убедиться в этом, надо записать электронные формулы всех трех элементов:
Na: 1s2 2s2 2p6 3s1
F: 1s2 2s2 2p5
Ne: 1s2 2s2 2p6
В электронных формулах нам важны только электронные конфигурации внешних уровней (они подчеркнуты). Запишем реакцию с помощью формул Льюиса:
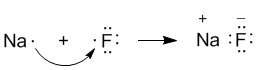
Натрий, отдав фтору свой 3s1-электрон, становится ионом Na+ и остается с заполненной 2s22p6 оболочкой, что отвечает электронной конфигурации атома неона. Точно такую же электронную конфигурацию приобретает атом F, приняв один электрон, отданный натрием. Теперь это ион F–. Разумеется, при этом ионы F– и Na+ продолжают оставаться все теми же элементами фтором и натрием, потому что никакие электронные переходы не могут изменить природу элемента – число протонов в его ядре.
Теперь в дополнение к ковалентной составляющей химической связи в молекуле Na+:F– добавляется еще и электростатическое притяжение между ионами натрия и фтора. Это увеличивает прочность химической связи. Однако ковалентная составляющая (стремление к октету) продолжает играть большую роль и в ионных соединениях.
Полярная ковалентная связь занимает промежуточное положение между чисто ковалентной связью и ионной связью. Так же, как и ионная, она может возникнуть между двумя атомами разных видов. В полярной ковалентной связи электроны смещаются от атома к атому не так сильно, как в ионной. Это происходит тогда, когда атомам энергетически невыгодно далеко “отпускать” свои собственные электроны, отданные в общую “копилку” – молекулярную орбиталь. Если электроны слишком далеко сдвинутся к одному из атомов, молекулярная орбиталь перестанет быть похожей на “октетную”. В то же время у разных атомов разные донорные и акцепторные свойства, поэтому связывающая электронная пара не располагается точно посередине между ядрами, как в ковалентной связи.
В качестве примера рассмотрим образование воды в реакции между атомами водорода (Z = 1) и кислорода (Z = 8). Для этого удобно сначала записать электронные формулы для внешних оболочек водорода (1s1) и кислорода (...2s2 2p4). Затем на помощь приходят формулы Льюиса, которые наглядно показывают, как образуются “завершенные” электронные оболочки рядом с атомами водорода и кислорода в молекуле воды:
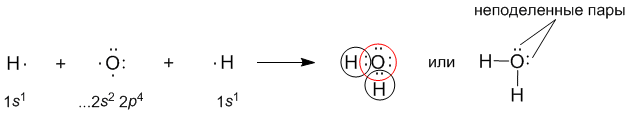
Оказывается, для этого необходимо взять именно два атома водорода на один атом кислорода. Однако природа такова, что акцепторные свойства атома кислорода выше, чем у атома водорода (о причинах этого чуть позже). Поэтому связывающие электронные пары в формуле Льюиса для воды слегка смещены к ядру атома кислорода. Связь в молекуле воды – полярная ковалентная, а на атомах появляются частичные положительные и отрицательные заряды.
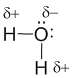
О том, как предсказать акцепторные свойства атомов и где проходит “граница” между полярной ковалентной и ионной связями мы поговорим в параграфе 3.4.
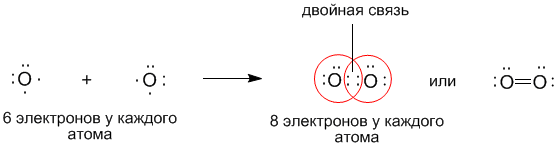
По теории Льюиса и правилу октета связь между атомами может осуществляться не обязательно одной, но и двумя и даже тремя поделенными парами, если этого требует правило октета. Такие связи называются двойными и тройными. Например, только что рассмотренный нами кислород может образовывать двухатомную молекулу с октетом электронов у каждого атома только тогда, когда между атомами помещаются две поделенные пары:
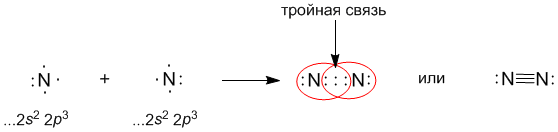
Атомы азота (...2s2 2p3 на последней оболочке) также связываются в двухатомную молекулу, но для организации октета электронов им требуется расположить между собой уже три поделенные пары:
В настоящее время принято изображать электронные пары (то есть химические связи) между атомами черточками. Каждая черточка – это поделенная пара электронов. В этом случае уже знакомые нам молекулы выглядят так:

Формулы с черточками между атомами называются структурными формулами. Чаще в структурных формулах не изображают неподеленные пары электронов, но в ряде случаев (мы столкнемся с ними при обсуждении донорно-акцепторных связей) неподеленные пары играют важную роль.
Структурные формулы очень хороши для изображения молекул: они четко показывают – как атомы связаны между собой, в каком порядке, какими связями.
Связывающая пара электронов в формулах Льюиса – то же самое, что одна черточка в структурных формулах.
Двойные и тройные связи имеют общее название – кратные связи. О молекуле азота говорят, что она имеет порядок связи, равный трем. В молекуле кислорода порядок связи равен двум. Порядок связи в молекулах водорода и хлора – один. У водорода и хлора уже не кратная, а простая связь.
Порядок связи – это число обобществленных поделенных пар между двумя связанными атомами. Порядок связи выше трех не встречается.
Таблица 3-1. Длины и прочности связей между атомами азота в различных соединениях.
Связь |
Длина связи в ангстремах 1А = 10–8 см |
Прочность связи в кДж на одинаковое число молекул |
Проcтая N–N |
1,45 |
58,5 |
Двойная N=N |
1,25 |
456 |
Тройная
|
1.098 |
945 |
** Рассмотрим данные по длинам и прочностям связей между атомами азота в различных его соединениях. В таблице 3-1 длины связей приведены в специальных единицах – ангстремах (1А = 10–8см). Относительную прочность связей можно оценить по энергии, которая необходима для разрыва связей между атомами азота в разных соединениях. Эта энергия дается для одинакового числа молекул таких соединений. Чем выше кратность связи, тем она короче и прочнее.
Чем выше порядок связи, тем прочнее связаны между собой атомы и тем короче сама связь.
Природа металлической связи. Физико-механические свойства кристаллических веществ, определяемые этим типом связи.
В узлах кристаллической решётки расположены положительные ионы металла. Между ними беспорядочно, подобно молекулам газа, движутся валентные электроны, происходящие из атомов металлов от атомов при образовании ионов. Эти электроны играют роль цемента, удерживая вместе положительные ионы; в противном случае решётка распалась бы под действием сил отталкивания между ионами. Вместе с тем и электроны удерживаются ионами в пределах кристаллической решётки и не могут её покинуть. Силы связи не локализованы и не направлены. Поэтому в большинстве случаев проявляются высокие координационные числа (например, 12 или 8).
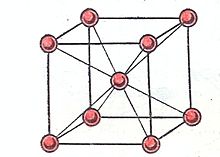
![]()
Рис.1.Расположение ионов в кристалле щелочного металла
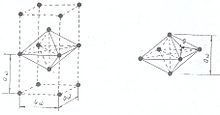
Рис.2.Связывающее звено кристаллической решётки щелочного металла
Так, щелочные металлы кристаллизуются в кубической объёмно-центрированной решётке, и каждый положительно заряженный ион щелочного металла имеет в кристалле по восемь ближайших соседей — положительно заряженных ионов щелочного металла (рис.1). Кулоновское отталкивание одноимённо-заряженных частиц (ионов) компенсируется электростатическим притяжением к электронам связывающих звеньев, имеющих форму искажённого сплющенного октаэдра — квадратной бипирамиды, высота которой и рёбра базиса равны величине постоянной трансляционной решётке aw кристалла щелочного металла (рис.2).
Связывающие электроны становятся общими для системы из шести положительных ионов щелочных металлов и удерживают последние от кулоновского отталкивания.
Величина постоянной трансляционной решётки aw кристалла щелочного металла значительно превышает длину ковалентной связи молекулы щелочного металла, поэтому принято считать, что электроны в металле находятся в свободном состоянии:
Щёлочный металл |
Li |
Na |
K |
Rb |
Cs |
Постоянная решётка aw,Å |
3,5021 |
4,2820 |
5,247 |
5,69 |
6,084 |
Длина ковалентной связи, Me2, Å |
2,67 |
3,08 |
3,92 |
4,10 |
4,30 |
Математическое построение, связанное со свойствами свободных электронов в металле, обычно отождествляют с «поверхностью Ферми», которую следует рассматривать как геометрическое место, где пребывают электроны, обеспечивая основное свойство металла — проводить электрический ток. Таким образом, электрический ток в металлах — это движение сорванных с орбитального радиуса электронов в поле положительно заряженных ионов, находящихся в узлах кристаллической решётки металла. Выход и вход свободных электронов в связывающее звено кристалла осуществляется через точки «0», равноудалённые от положительных ионов атомов (рис.2).
Свободное движение электронов в металле подтверждено в 1916 году опытом Толмена и Стюарта по резкому торможению быстро вращающейся катушки с проводом — свободные электроны продолжали двигаться по инерции, в результате чего гальванометр регистрировал импульс электрического тока. Свободное движение электронов в металле обусловливает высокую теплопроводность металла и склонность металлов к термоэлектронной эмиссии, происходящей при умеренной температуре.
Колебания ионов кристаллической решётки создаёт сопротивление движению электронов по металлу, сопровождающееся разогревом металла. В настоящее время важнейшим признаком металлов считается отрицательный температурный коэффициент электрической проводимости, то есть понижение проводимости с ростом температуры. С понижением температуры электросопротивление металлов уменьшается, вследствие уменьшения колебаний ионов в кристаллической решётке. В процессе исследования свойств материи при низких температурах Камерлинг-Оннес открывает явление сверхпроводимости. В 1911 году ему удаётся обнаружить уменьшение электросопротивления ртути при температуре кипения жидкого гелия (4,2 К) до нуля. В 1913 году Камерлинг-Оннесу присуждается Нобелевская премия по физике со следующей формулировкой: «За исследование свойств веществ при низких температурах, которые привели к производству жидкого гелия.»
Однако теория сверхпроводимости была создана позднее. В её основе лежит концепция куперовской электронной пары — коррелированного состояния связывающих электронов с противоположными спинамии и импульсами, и, следовательно, сверхпроводимость можно рассматривать как сверхтекучесть электронного газа, состоящего из куперовских пар электронов, через ионную кристаллическую решётку. В 1972 году авторам теории БКШ — Бардину, Куперу и Шрифферу присуждена Нобелевская премия по физике «За создание теории сверхпроводимости, обычно называемой БКШ-теорией».
Ионная связь. Электроотрицательность элемента. Водородная связь. Межмолекулярные взаимодействия.
характер химической связи часто находит отражение в агрегатном состоянии и физических свойствах вещества. Такие ионные соединения, как хлорид натрия NaCl твердые и тугоплавкие потому, что между зарядами их ионов "+" и "–" существуют мощные силы электростатического притяжения.
Отрицательно заряженный ион хлора притягивает не только "свой" ион Na+, но и другие ионы натрия вокруг себя. Это приводит к тому, что около любого из ионов находится не один ион с противоположным знаком, а несколько (рис. 3-12).
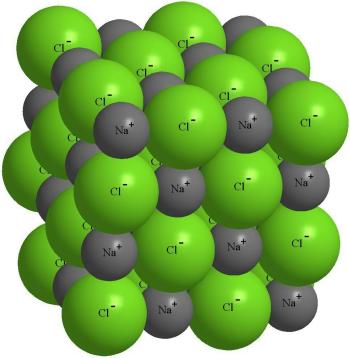
Рис. 3-12. Строение кристалла поваренной соли NaCl.
Фактически, около каждого иона хлора располагается 6 ионов натрия, а около каждого иона натрия - 6 ионов хлора. Такая упорядоченная упаковка ионов называется ионным кристаллом. Если в кристалле выделить отдельный атом хлора, то среди окружающих его атомов натрия уже невозможно найти тот, с которым хлор вступал в реакцию. Притянутые друг к другу электростатическими силами, ионы крайне неохотно меняют свое местоположение под влиянием внешнего усилия или повышения температуры. Но если хлорид натрия расплавить и продолжать нагревать в вакууме, то он испаряется, образуя двухатомные молекулы NaCl . Это говорит о том, что силы ковалентного связывания никогда не выключаются полностью.
По-другому устроены металлические кристаллы. Если рассмотреть кусочек металлического натрия, то обнаружится, что внешне он сильно отличается от поваренной соли. Натрий - мягкий металл, легко режется ножом, расплющивается молотком, его можно без труда расплавить в чашечке на спиртовке (температура плавления 97,8 оС). В кристалле натрия каждый атом окружен восемью другими такими же атомами (рис. 3-13).
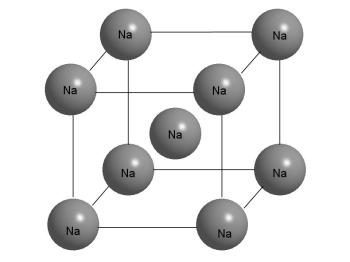
Рис. 3-13. Строение кристалла металлического Na. Из рисунка видно, что атом Na в центре куба имеет 8 ближайших соседей. Но это же можно сказать и о любом другом атоме в кристалле, поскольку все они одинаковы. Кристалл состоит из "бесконечно" повторяющихся фрагментов, изображенных на этом рисунке.
На рис. 3-13 металлический кристалл выглядит достаточно простым, но на самом деле его электронное устройство сложнее, чем у кристаллов ионных солей. На внешней электронной оболочке элементов-металлов недостаточно электронов для образования полноценной "октетной" ковалентной или ионной связи. Поэтому в газообразном состоянии большинство металлов состоит из одноатомных молекул, (т.е. отдельных, не связанных между собой атомов). Типичный пример - пары ртути. Таким образом, металлическая связь между атомами металлов возникает только в жидком и твердом агрегатном состоянии.
Описать металлическую связь можно следующим образом: часть атомов металла в образующемся кристалле отдают в пространство между атомами свои валентные электроны (у натрия это ...3s1), превращаясь в ионы (рис. 3-14). Поскольку все атомы металла в кристалле одинаковы, каждый из них имеет равные с другими шансы потерять валентный электрон. Иными словами, переход электронов между нейтральными и ионизированными атомами металла происходит без затрат энергии. Часть электронов при этом всегда оказывается в пространстве между атомами в виде "электронного газа". Эти свободные электроны, во-первых, удерживают атомы металла на определенном равновесном расстоянии друг от друга. Во-вторых, они придают металлам характерный "металлический блеск" (свободные электроны могут взаимодействовать с квантами света). В-третьих, свободные электроны обеспечивают металлам хорошую электропроводность. Высокая теплопроводность металлов тоже объясняется наличием свободных электронов в межатомном пространстве - они легко "откликаются" на изменения энергии и способствуют ее быстрому переносу в кристалле.
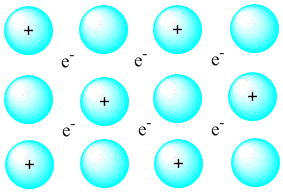
Рис. 3-14. Упрощенная модель электронного строения металлического кристалла.
** На примере металла натрия рассмотрим природу металлической связи с точки зрения представлений об атомных орбиталях. У атома натрия, как и у многих других металлов, имеется недостаток валентных электронов, зато имеются свободные валентные орбитали. Единственный 3s-электрон натрия способен перемещаться на любую из свободных и близких по энергии соседних орбиталей. При сближении атомов в кристалле внешние орбитали соседних атомов перекрываются, благодаря чему отданные электроны свободно перемещаются по всему кристаллу.
О
 днако
"электронный газ" вовсе не
беспорядочен, как может показаться.
Свободные электроны в металлическом
кристалле находятся на перекрывающихся
орбиталях и в какой-то мере обобществляются,
образуя подобие ковалентных связей. У
натрия, калия, рубидия и других
металлических s-элементов обобществленных
электронов просто мало, поэтому их
кристаллы непрочные и легкоплавкие. С
увеличением числа валентных электронов
прочность металлов, как правило,
возрастает. В конце параграфа мы еще
вернемся к этому вопросу.
днако
"электронный газ" вовсе не
беспорядочен, как может показаться.
Свободные электроны в металлическом
кристалле находятся на перекрывающихся
орбиталях и в какой-то мере обобществляются,
образуя подобие ковалентных связей. У
натрия, калия, рубидия и других
металлических s-элементов обобществленных
электронов просто мало, поэтому их
кристаллы непрочные и легкоплавкие. С
увеличением числа валентных электронов
прочность металлов, как правило,
возрастает. В конце параграфа мы еще
вернемся к этому вопросу.
Таким образом, металлическую связь склонны образовывать элементы, атомы которых на внешних оболочках имеют мало валентных электронов. Эти валентные электроны, осуществляющие металлическую связь, обобществлены настолько, что могут перемещаться по всему металлическому кристаллу и обеспечивают высокую электропроводность металла.
Кристалл NaCl не проводит электрический ток, потому что в пространстве между ионами нет свободных электронов. Все электроны, отданные атомами натрия, прочно удерживают около себя ионы хлора. В этом одно из существенных отличий ионных кристаллов от металлических.
То, что вы теперь знаете о металлической связи, позволяет объяснить и высокую ковкость (пластичность) большинства металлов. Металл можно расплющить в тонкий лист, вытянуть в проволоку. Дело в том, что отдельные слои из атомов в кристалле металла могут относительно легко скользить один по другому: подвижный "электронный газ" постоянно смягчает перемещение отдельных положительных ионов, экранируя их друг от друга (рис. 3-14).
Разумеется, ничего подобного нельзя сделать с поваренной солью, хотя соль - тоже кристаллическое вещество. В ионных кристаллах валентные электроны прочно связаны с ядром атома (рис. 3-12). Сдвиг одного слоя ионов относительно другого приводит к сближению ионов одинакового заряда (рис. 3-15) и вызывает сильное отталкивание между ними, в результате чего происходит разрушение кристалла (NaCl - хрупкое вещество).
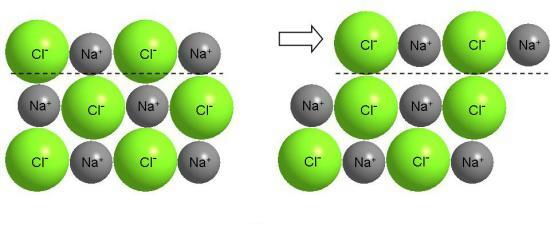
Рис. 3-15. Сдвиг слоев ионного кристалла вызывает появление больших сил отталкивания между одноименными ионами и разрушение кристалла.
Еще один вид кристаллов - это так называемые молекулярные кристаллы. "Строительными деталями" в них выступают отдельные молекулы, которые удерживаются рядом друг с другом силами межмолекулярного взаимодействия.
Т акие силы могут иметь различную природу – например, у воды это водородные связи (мы еще вернемся к ним в §7.4). Молекулы также могут связываться друг с другом слабым электростатическим диполь-дипольным взаимодействием – т.е. притяжением разноименных частичных зарядов (как в кристаллических органических веществах). Это могут быть и межмолекулярные силы, возникающие при поляризации одних молекул другими - в результате случайного перераспределения электронной плотности в одной из молекул. Такое взаимодействие называется индукционным или наведенным. Например, молекулы I2 неполярные, но состоят из "больших" (относительно, конечно) атомов, электронные оболочки которых легко поляризуются под влиянием соседних частиц или стенок сосуда. Силы межмолекулярного взаимодействия слабее настоящих химических связей, поэтому молекулярные кристаллы непрочные. В принципе, эти же силы удерживают молекулы друг около друга и в жидкостях и молекулярные кристаллы обычно легко плавятся или даже возгоняются. Возгонка – переход вещества из твердого состояния в газообразное, минуя жидкое (посмотрите видеоопыт "возгонка йода" из Единой коллекции цифровых образовательных ресурсов).
Но если температура не велика (ниже точки плавления или возгонки), то молекулы собираются в упорядоченные каркасы, где каждая из них ориентирована в пространстве строго определенным образом. Хороший пример - молекулярные кристаллы льда, в которых молекулы H2O располагаются в строгом порядке относительно друг друга (рис. 3-16)
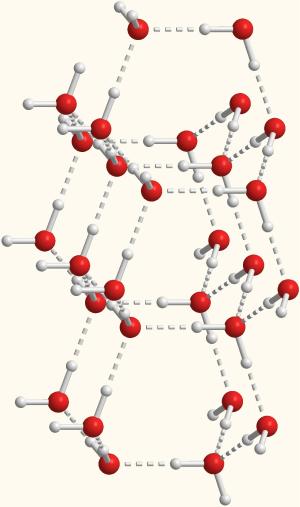
Рис. 3-16. Модель растущего кристалла льда (красные шарики - атомы кислорода, белые шарики - атомы водорода в молекулах H2O). Отдельные молекулы воды связаны друг с другом водородными связями (показаны пунктирными линиями) в строгом порядке. Это приводит к причудливым симметричным формам кристаллов (вспомните форму снежинок). Лёд - молекулярный кристалл.
Наконец, существуют очень прочные кристаллы, в которых атомы в решетке удерживаются прочными ковалентными связями. Такие кристаллы называются ковалентными каркасными кристаллами или атомными кристаллами. Мельчайшими структурными частицами в них являются отдельные атомы, связанные ковалентными или полярными ковалентными связями в «бесконечный» трехмерный каркас. Благодаря этому вещества с атомной кристаллической решеткой обладают высокой механической прочностью, не имеют запаха и нерастворимы в воде.
На рис. 3-17 изображен фрагмент атомной кристаллической решетки оксида кремния SiO2 (песок, кварц). Атомы кремния и кислорода здесь связаны между собой прочными полярными ковалентными связями. Как мы видим, атомы связаны только простыми связями и структура SiO2 совсем не такая, как у углекислого газа СO2, который состоит из отдельных молекул O=C=O с двойными связями. Кремний не склонен образовывать двойные (и вообще кратные) связи. Вероятно, немалую роль в этом играет относительно большой размер атомов кремния. Вы уже знаете, что двойные связи более короткие, чем простые, и хорошо "работают" только на малых расстояниях между ядрами. Большой диаметр атомов кремния препятствует образованию кратных связей.
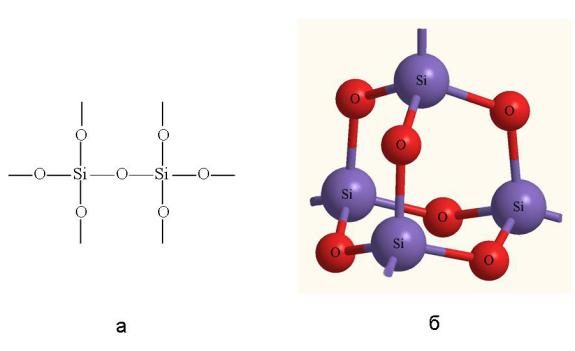
Рис. 3-17. Оксид кремния SiO2 – фрагмент структурной формулы (а) и строение атомного кристалла (б).
** Если бы SiO2 был устроен так же, как СO2, то был бы, скорее всего, газообразным. В этом случае мы лишились бы не только песчаных пляжей, стекла, керамики, многих драгоценных камней (таких как горный хрусталь и аметист), но и бетонных сооружений и еще множества строительных материалов, в состав которых входит оксид кремния. Кроме того, с поверхности Земли исчезло бы огромное количество силикатных минералов. Даже трудно представить последствия, которые имело бы небольшое уменьшение линейных размеров атомов кремния!
Еще один очень известный атомный кристалл - это алмаз (рис. 3-18). Он состоит только из одного элемента углерода - того же самого, из которого состоит обыкновенная сажа и графит. В алмазе каждый 4-х валентный атом углерода связан с другим атомом углерода ковалентной связью и количество таких связанных в каркас атомов чрезвычайно велико. Алмаз можно было бы назвать гигантской молекулой, если бы к молекулам не предъявлялось требование иметь постоянный состав.
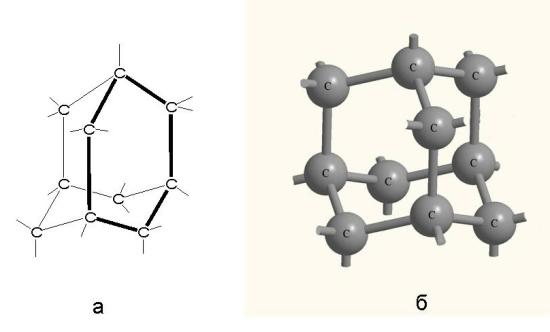
Рис. 3-18. Структурная формула алмаза (а) и строение его атомного кристалла (б). Атомы углерода образуют изогнутые шестичленные кольца. При этом каждый атом углерода находится в центре правильного тетраэдра, в вершинах которого - другие углеродные атомы. связанные с ним ковалентными связями. Углы между связями составляют 109о - как в математическом тетраэдре.
В кристалле графита атомы углерода связаны по-иному (рис. 3-19). Они объединены в слои, состоящие из плоских шестиугольников. В первом приближении можно представить, что в этих шестиугольниках атомы углерода связаны между собой чередующимися простыми и двойными связями. Расстояние между отдельными слоями в графите довольно велико, а силы взаимодействия между ними довольно слабы (в основном это слабые межмолекулярные связи, показанные вертикальными пунктирными линиями), поэтому графит может расщепляться на тонкие чешуйки. При легком нажатии чешуйки легко прилипают к бумаге – вот почему из графита делают грифели карандашей. Графит и алмаз очень несхожи по своим свойствам, хотя состоят из одного и того же элемента - углерода.
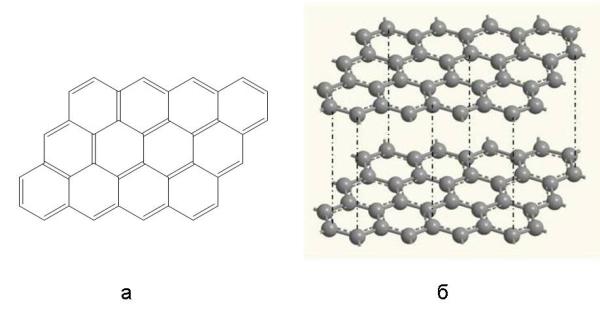
Рис. 3-19. Структурная формула участка одного углеродного слоя графита (а) и пространственное расположение атомов в кристаллической решетке графита (б). Внутри слоев атомы углерода связаны друг с другом прочными ковалентными связями, а между слоями действуют гораздо более слабые силы межмолекулярного взаимодействия.
Вы можете провести простой опыт, когда в очередной раз будете точить карандаш. Разотрите между пальцами немного грифельного порошка - на ощупь он будет жирным. Капельки воды не смачивают графит - проверьте это на испачканных графитом пальцах. Графит и алмаз - ближайшие родственники, хотя и обладают разными свойствами.
Графит - непрочное вещество, его легко превратить в порошок. Совсем другие свойства проявляет алмаз. Он настолько тверд, что оставляет царапины на большинстве материалов. Алмаз проверяют на подлинность, царапая им стекло. Другой метод определения подлинности алмаза таков: если напылить на грань алмаза мелкие капли воды, то они не растекаются по грани, потому что неполярный ковалентный алмаз, как и графит, не притягивает полярные молекулы воды. Все поддельные (не путайте с искусственными!) алмазы можно сделать только из соединений с полярными ковалентными связями. По их поверхности вода растекается так же легко, как по чистому стеклу.
Когда химический элемент образует два или больше простых веществ, различных по строению и свойствам, такое явление называется аллотропией. Графит и алмаз - две аллотропные модификации углерода. Аллотропные модификации при определенных условиях могут переходить друг в друга. Например, при очень высоких давлениях и температурах графит может переходить в алмаз. Именно так из графита делают искусственные алмазы.
Искусственные алмазы не пригодны для ювелирных изделий - они мелкие и черного цвета. Но для промышленных нужд такие алмазы являются очень ценным сырьем. Пылевидный алмазный порошок можно получить и взрывом (в закрытой камере) специальных взрывчатых веществ, содержащих углерод. Возникновение природных алмазов в недрах Земли, вероятно, тоже происходит под влиянием огромных температур и давлений, но в течение неизмеримо более длительного времени.
Термодинамические функции состояния системы. Термодинамические законы и расчёты.
Термодинамическая функция состояния — в термодинамике некая функция, зависящая от нескольких независимых параметров, которые однозначно определяют состояние термодинамической системы. Значение термодинамической функции состояния зависит только от состояния термодинамической системы и не зависит от того, как система пришла в это состояние. Частным случаем функций состояний являются термодинамические потенциалы.
Термодинамическими величинами называют физические величины, применяемые при описании состояний и процессов в термодинамических системах.
Термодинамика рассматривает эти величины как некоторые макроскопические параметры и функции, присущие системе, но не связанные с её микроскопическим устройством. Вопросы микроскопического устройства изучает статистическая физика.
Функции состояния
Функции состояния зависят только от текущего состояния системы и не зависят от пути, по которому система пришла в это состояние.
Функции состояния в термодинамике включают:
температуру,
давление,
объём,
энтропию,
а также термодинамические потенциалы.
В зависимости от выбранной модели некоторые из этих величин, строго говоря, могут быть не функциями, а независимыми переменными.
Эти величины не являются независимыми. Связь между термодинамическими параметрами для конкретной системы называется уравнением состояния.
В случае, если известно каноническое уравнение состояния, задание любой пары параметров из следующих:
энтропия и объём,
энтропия и давление,
температура и объём,
температура и давление,
позволяет вычислить остальные два параметра.
II закон термодинамики. Характеристические функции системы. Уравнение энергетического баланса системы, его анализ.
Второе начало термодинамики — физический принцип, накладывающий ограничение на направление процессов передачи тепла между телами.
Второе начало термодинамики запрещает так называемые вечные двигатели второго рода, показывая, что коэффициент полезного действия не может равняться единице, поскольку для кругового процесса температура холодильника не может равняться абсолютному нулю (невозможно построить замкнутый цикл, проходящий через точку с нулевой температурой).
Второе начало термодинамики является постулатом, не доказываемым в рамках термодинамики. Оно было создано на основе обобщения опытных фактов и получило многочисленные экспериментальные подтверждения.
уществуют несколько эквивалентных формулировок второго начала термодинамики:
Постулат Клаузиуса: «Невозможен процесс, единственным результатом которого являлась бы передача тепла от более холодного тела к более горячему» (такой процесс называется процессом Клаузиуса).
Постулат Томсона (Кельвина): «Невозможен круговой процесс, единственным результатом которого было бы производство работы за счет охлаждения теплового резервуара» (такой процесс называется процессом Томсона).
Эквивалентность
этих формулировок легко показать. В
самом деле, допустим, что постулат
Клаузиуса неверен, то есть существует
процесс, единственным результатом
которого была бы передача тепла от более
холодного тела к более горячему. Тогда
возьмем два тела с различной температурой
(нагреватель и холодильник) и проведем
несколько циклов тепловой
машины, забрав тепло
![]() у
нагревателя, отдав
у
нагревателя, отдав
![]() холодильнику
и совершив при этом работу
холодильнику
и совершив при этом работу
![]() .
После этого воспользуемся процессом
Клаузиуса и вернем тепло
от
холодильника нагревателю. В результате
получается, что мы совершили работу
только за счет отъёма теплоты от
нагревателя, то есть постулат Томсона
тоже неверен.
.
После этого воспользуемся процессом
Клаузиуса и вернем тепло
от
холодильника нагревателю. В результате
получается, что мы совершили работу
только за счет отъёма теплоты от
нагревателя, то есть постулат Томсона
тоже неверен.
С другой стороны, предположим, что неверен постулат Томсона. Тогда можно отнять часть тепла у более холодного тела и превратить в механическую работу. Эту работу можно превратить в тепло, например, с помощью трения, нагрев более горячее тело. Значит, из неверности постулата Томсона следует неверность постулата Клаузиуса.
Таким образом, постулаты Клаузиуса и Томсона эквивалентны.
Другая формулировка второго начала термодинамики основывается на понятии энтропии:
«Энтропия изолированной системы не может уменьшаться» (закон неубывания энтропии).
Такая формулировка основывается на представлении об энтропии как о функции состояния системы, что также должно быть постулировано.
Второе начало термодинамики в аксиоматической формулировке Рудольфа Юлиуса Клаузиуса (R. J. Clausius, 1865) имеет следующий вид:
Для
любой квазиравновесной термодинамической
системы существует однозначная функция
термодинамического состояния
![]() ,
называемая энтропией, такая, что ее
полный дифференциал
,
называемая энтропией, такая, что ее
полный дифференциал
![]() .
.
В состоянии с максимальной энтропией макроскопические необратимые процессы (а процесс передачи тепла всегда является необратимым из-за постулата Клаузиуса) невозможны.
Условия самопроизвольного протекания и предела протекания процесса. Термодинамика фазовых переходов.
Поведение всякой термодинамической системы в общем случае определяется одновременным действием двух факторов – энтальпийного, отражающего стремление системы к минимуму тепловой энергии, и энтропийного, отражающего противоположную тенденцию – стремление системы к максимальной неупорядоченности. Если для изолированных систем (ΔН = 0) направление и предел самопроизвольного протекания процесса однозначно определяется величиной изменения энтропии системы ΔS, а для систем, находящихся при температурах, близких к абсолютному нулю (S = 0 либо S = const) критерием направленности самопроизвольного процесса является изменение энтальпии ΔН, то для закрытых систем при температурах, не равных нулю, необходимо одновременно учитывать оба фактора. Направление и предел самопроизвольного протекания процесса в любых системах определяет более общий принцип минимума свободной энергии:
Самопроизвольно могут протекать только те процессы, которые приводят к понижению свободной энергии системы; система приходит в состояние равновесия, когда свободная энергия достигает минимального значения.
Для закрытых систем, находящихся в изобарно-изотермических либо изохорно-изотермических условиях свободная энергия принимает вид изобарно-изотермического либо изохорно-изотермического потенциалов (т.н. свободная энергия Гиббса и Гельмгольца соответственно). Данные функции называют иногда просто термодинамическими потенциалами, что не вполне строго, поскольку термодинамическими потенциалами являются также внутренняя энергия (изохорно-изэнтропный) и энтальпия (изобарно-изэнтропный потенциал).
Рассмотрим закрытую систему, в которой осуществляется равновесный процесс при постоянных температуре и объеме. Выразим работу данного процесса, которую обозначим Amax (поскольку работа процесса, проводимого равновесно, максимальна), из уравнений (I.53, I.54):
![]() (I.68)
(I.68)
![]() (I.69)
(I.69)
Преобразуем выражение (I.69), сгруппировав члены с одинаковыми индексами:
![]() (I.70)
(I.70)
Введя обозначение:
![]() (I.71)
(I.71)
получаем:
![]() (I.72)
(I.72)
![]() (I.73)
(I.73)
Функция есть изохорно-изотермический потенциал (свободная энергия Гельмгольца), определяющий направление и предел самопроизвольного протекания процесса в закрытой системе, находящейся в изохорно-изотермических условиях.
Закрытую систему, находящуюся в изобарно-изотермических условиях, характеризует изобарно-изотермический потенциал G:
![]() (I.74)
(I.74)
![]() (I.75)
(I.75)
Поскольку –ΔF = Amax, можно записать:
![]() (I.76)
(I.76)
Величину А'max называют максимальной полезной работой (максимальная работа за вычетом работы расширения). Основываясь на принципе минимума свободной энергии, можно сформулировать условия самопроизвольного протекания процесса в закрытых системах.
Условия самопроизвольного протекания процессов в закрытых системах:
Изобарно-изотермические (P = const, T = const):
ΔG < 0, dG < 0
Скорость химической реакции, влияние концентрационного фактора на скорость. Кинетическое уравнение процесса.
Скорость химической реакции зависит от природы реагирующих веществ и условий протекания реакции: концентрации с, температуры t , присутствия катализаторов, а также от некоторых других факторов (например, от давления - для газовых реакций, от измельчения - для твердых веществ, от радиоактивного облучения).
Влияние концентраций реагирующих веществ. Чтобы осуществлялось химическое взаимодействие веществ А и В, их молекулы (частицы) должны столкнуться. Чем больше столкновений, тем быстрее протекает реакция. Число же столкновений тем больше, чем выше концентрация реагирующих веществ. Отсюда на основе обширного экспериментального материала сформулирован основной закон химической кинетики, устанавливающий зависимость скорости реакции от концентрации реагирующих веществ:
Cкорость химической реакции пропорциональна произведению концентраций реагирующих веществ.
Для реакции ( I ) этот закон выразится уравнением
v = kcA cB , (1)
где сА и сВ - концентрации веществ А и В, моль/л; k - коэффициент пропорциональности, называемый константой скорости реакции. Основной закон химической кинетики часто называют законом действующих масс.
Из уравнения (1) нетрудно установить физический смысл константы скорости k : она численно равна скорости реакции, когда концентрации каждого из реагирующих веществ составляют 1 моль/л или когда их произведение равно единице.
Константа скорости реакции k зависит от природы реагирующих веществ и от температуры, но не зависит от их концентраций.
Уравнение (1), связывающее скорость реакции с концентрацией реагирующих веществ, называется кинетическим уравнением реакции. Если опытным путем определено кинетическое уравнение реакции, то с его помощью можно вычислять скорости при других концентрациях тех же реагирующих веществ.
Температурная зависимость скорости процесса. Правило Вант-Гоффа, уравнение Аррениуса, энергия активации. Катализ.
Зависимость скорости реакции от температуры определяется правилом Вант-Гоффа:
При повышении температуры на каждые 10о скорость большинства реакций увеличивается в 2-4 раза.
Математически эта зависимость выражается соотношением
![]()
vt 2 = vt 1 γ ,
где vt 1 , vt 2 - скорости реакции соответственно при начальной ( t 1 ) и конечной ( t 2 ) температурах, а γ - температурный коэффициент скорости реакции, который показывает, во сколько раз увеличивается скорость реакции с повышением температуры реагирующих веществ на 10°.
Правило Вант-Гоффа является приближенным и применимо лишь для ориентировочной оценки влияния температуры на скорость реакции. Температура влияет на скорость химической реакции, увеличивая константу скорости.
Уравнение Аррениуса.
где k – константа скорости реакции при температуре Т;
А – предэкспоненциальный множитель (коэффициент Аррениуса), учитывающий частоту столкновений частиц, ориентированных определенным образом.
e – Неперово число
EA – энергия активации реакции, Дж/моль
R – универсальная газовая постоянная (8,314 Дж/моль·К)
Т – абсолютная температура, К
Энергия активации.
Энергия активации – минимальная энергия взаимодействующих частиц, достаточная для того, чтобы все частицы вступили в химическую реакцию.
Автоколебательные кинетические процессы в биологических системах.
Циклические изменения концентраций промежуточных соединений, и в первую очередь вещества Y, будут влиять на скорость образования конечного продукта Р, концентрация которого в системе также будет изменяться периодически во времени: то нарастать, то падать. Такой кинетический режим называется автоколебательным. Причем, если концентрация исходного вещества А в системе поддерживается постоянной (стационарное состояние), то автоколебательный режим устойчив во времени, а если с(А) уменьшается во времени, то автоколебательный режим носит затухающий характер.
Теория гомогенного и гетерогенного катализа.
Если катализатор и реагент, участвующие в процессе, находятся в одной фазе, то такой процесс называют гомогенным, а если катализатор и реагент находятся в разных фазах, - гетерогенным.
Химическое равновесие, закон действующих масс. Смещение равновесия, принцип Ле – Шателье. Уравнение химического сродства.
Если система находится в состоянии равновесия, то она будет пребывать в нем до тех пор, пока внешние условия сохраняются постоянными. Если же условия изменятся, то система выйдет из равновесия — скорости прямого и обратного процессов изменятся неодинаково — будет протекать реакция. Наибольшее значение имеют случаи нарушения равновесия вследствие изменения концентрации какого-либо из веществ, участвующих в равновесии, давления или температуры.
Рассмотрим каждый из этих случаев.
Нарушение
равновесия вследствие изменения
концентрации какого-либо из веществ,
участвующих в реакции. Пусть водород,
иодоводород и пары иода находятся в
равновесии друг с другом при определенных
температуре и давлении. Введем в систему
дополнительно некоторое количество
водорода. Согласно закону действия
масс, увеличение концентрации водорода
повлечет за собой увеличение скорости
прямой реакции — реакции синтеза HI,
тогда как скорость обратной реакции не
изменится. В прямом направлении реакция
будет теперь протекать быстрее, чем в
обратном. В результате этого концентрации
водорода и паров иода будут уменьшаться,
что повлечет за собою замедление прямой
реакции, а концентрация HI будет возрастать,
что вызовет ускорение обратной реакции.
Через некоторое время скорости прямой
и обратной реакций вновь сравняются—
установится новое равновесие. Но при
этом концентрация HI будет теперь выше,
чем она была до добавления
![]() ,
а концентрация
,
а концентрация
![]() —
ниже.
—
ниже.
Процесс
изменения концентраций, вызванный
нарушением равновесия, называется
смещением или сдвигом равновесия. Если
при этом происходит увеличение
концентраций веществ, стоящих в правой
части уравнения (и, конечно, одновременно
уменьшение концентраций веществ, стоящих
слева), то говорят, что равновесие
смещается вправо, т. е. в направлении
течения прямой реакции; при обратном
изменении концентраций говорят о
смещении равновесия влево — в направлении
обратной реакции. В рассмотренном
примере равновесие сместилось вправо.
При этом то вещество
![]() ,
увеличение концентрации которого
вызвало нарушение равновесия, вступило
в реакцию — его концентрация понизилась.
,
увеличение концентрации которого
вызвало нарушение равновесия, вступило
в реакцию — его концентрация понизилась.
Таким образом, при увеличении концентрации какого-либо из веществ, участвующих в равновесии, равновесие смещается в сторону расхода этого вещества; при уменьшении концентрации какого-либо из веществ равновесие смещается в сторону образования этого вещества.
Нарушение равновесия вследствие изменения давления (путем уменьшения или увеличения объема системы). Когда в реакции участвуют газы, равновесие может нарушиться при изменении объема системы.
Рассмотрим влияние давления на реакцию между монооксидом азота и кислородом:
![]()
Пусть
смесь газов
![]() ,
,
![]() и
и
![]() находится
в химическом равновесии при определенной
температуре и давлении. Не изменяя
температуры, увеличим давление так,
чтобы объем системы уменьшился в 2 раза.
В первый момент парциальные давления
и концентрации всех газов возрастут
вдвое, но при этом изменится соотношение
между скоростями прямой и обратной
реакций — равновесие нарушится.
находится
в химическом равновесии при определенной
температуре и давлении. Не изменяя
температуры, увеличим давление так,
чтобы объем системы уменьшился в 2 раза.
В первый момент парциальные давления
и концентрации всех газов возрастут
вдвое, но при этом изменится соотношение
между скоростями прямой и обратной
реакций — равновесие нарушится.
В
самом деле, до увеличения давления
концентрации газов имели равновесные
значения
![]() ,
,
![]() и
и
![]() ,
а скорости прямой и обратной реакций
были одинаковы и определялись уравнениями:
,
а скорости прямой и обратной реакций
были одинаковы и определялись уравнениями:
![]()
В
первый момент после сжатия концентрации
газов увеличатся вдвое по сравнению с
их исходными значениями и будут равны
соответственно
![]() ,
,
![]() и
и
![]() .
При этом скорости прямой и обратной
реакций будут определяться уравнениями:
.
При этом скорости прямой и обратной
реакций будут определяться уравнениями:
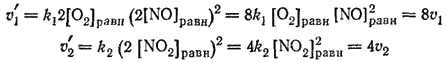
Таким
образом, в результате увеличения давления
скорость прямой реакции возросла в 8
раз, а обратной — только в 4 раза.
Равновесие в системе нарушится — прямая
реакция будет преобладать над обратной.
После того как скорости сравняются,
вновь установится равновесие, но
количество
![]() в
системе возрастет, равновесие сместится
вправо.
в
системе возрастет, равновесие сместится
вправо.
Нетрудно видеть, что неодинаковое изменение скоростей прямой и обратной реакций связано с тем, что в левой и в правой частях уравнения рассматриваемой реакции различно число молекул газов: одна молекула кислорода и две молекулы монооксида азота (всего три молекулы газов) превращаются в две молекулы газа — диоксида азота. Давление газа есть результат ударов его молекул о стенки сосуда; при прочих равных условиях давление газа тем выше, чем больше молекул заключено в данном объеме газа. Поэтому реакция, протекающая с увеличением числа молекул газов, приводит к возрастанию давления, а реакция, протекающая с уменьшением числа молекул газов, — к его понижению.
Коллигативные свойства растворов не электролитов. Закон Рауля, явление осмоса, уравнение Вант – Гоффа.
Коллигативные свойства растворов — это те свойства, которые при данных условиях оказываются равными и независимыми от химической природы растворённого вещества; свойства растворов, которые зависят лишь от количества кинетических единиц и от их теплового движения.
В этой статье будут кратко рассмотрены изменения термодинамических свойств растворов относительно свойств растворителя:
понижение давления пара,
повышение температуры кипения,
понижение температуры замерзания,
осмотическое давление.
Основные статьи на данные темы можно найти по вышеуказанным ссылкам.
Примечание: идеальным называют раствор, образование которого не сопровождается химическим взаимодействием, изменением объёма и тепловым эффектом.
Ещё одно примечание: все формулы в статье даны для растворов неэлектролитов. При рассмотрении растворов электролитов необходимо умножать правую часть формулы на изотонический коэффициент (i).
Законы Рауля — общее название открытых французским химиком Ф. М. Раулем в 1887 г. количественных закономерностей, описывающих некоторые коллигативные (зависящие от концентрации, но не от природы растворённого вещества) свойства растворов.
Первый закон Рауля связывает давление насыщенного пара над раствором с его составом; он формулируется следующим образом:
Парциальное давление насыщенного пара компонента раствора прямо пропорционально его мольной доле в растворе, причём коэффициент пропорциональности равен давлению насыщенного пара над чистым компонентом.
![]()
Для бинарного раствора, состоящего из компонентов А и В (компонент А считаем растворителем) удобнее использовать другую формулировку:
Относительное понижение парциального давления пара растворителя над раствором не зависит от природы растворённого вещества и равно его мольной доле в растворе.
![]()

На поверхности оказывается меньше способных испаряться молекул растворителя, ведь часть места занимает растворённое вещество.
Растворы, для которых выполняется закон Рауля, называются идеальными. Идеальными при любых концентрациях являются растворы, компоненты которых очень близки по физическим и химическим свойствам (оптические изомеры, гомологи и т. п.), и образование которых не сопровождается изменением объёма и выделением либо поглощением теплоты. В этом случае силы межмолекулярного взаимодействия между однородными и разнородными частицами примерно одинаковы, и образование раствора обусловлено лишь энтропийным фактором.
Растворы, компоненты которых существенно различаются по физическим и химическим свойствам, подчиняются закону Рауля лишь в области очень малых концентраций; при больших концентрациях наблюдаются отклонения от закона Рауля. Случай, когда истинные парциальные давления паров над смесью больше, чем вычисленные по закону Рауля, называют положительными отклонениями. Противоположный случай, когда парциальные давления паров компонентов оказываются меньше вычисленных — отрицательные отклонения.
Причиной отклонений от закона Рауля является то обстоятельство, что однородные частицы взаимодействуют друг с другом иначе, чем разнородные (сильнее в случае положительных и слабее в случае отрицательных отклонений).
Реальные растворы с положительными отклонениями от закона Рауля образуются из чистых компонентов с поглощением теплоты (ΔНраств > 0); объём раствора оказывается больше, чем сумма исходных объёмов компонентов (ΔV > 0). Растворы с отрицательными отклонениями от закона Рауля образуются с выделением теплоты (ΔНраств < 0); объём раствора в этом случае будет меньше, чем сумма исходных объёмов компонентов (ΔV < 0).
Второй закон Рауля
Тот факт, что давление паров над раствором отличается от давления паров над чистым растворителем, существенно влияет на процессы кристаллизации и кипения. Из первого закона Рауля выводятся два следствия, касающиеся понижения температуры замерзания и повышения температуры кипения растворов, которые в объединённом виде известны как второй закон Рауля.
Условием кристаллизации является равенство давления насыщенного пара растворителя над раствором давлению пара над твёрдым растворителем. Поскольку давление пара растворителя над раствором всегда ниже, чем над чистым растворителем, это равенство всегда будет достигаться при температуре более низкой, чем температура замерзания растворителя. Так, океанская вода начинает замерзать при температуре около минус 2 °C.
Разность между температурой кристаллизации растворителя T°fr и температурой начала кристаллизации раствора Tfr есть понижение температуры кристаллизации.
Понижение температуры кристаллизации бесконечно разбавленных растворов не зависит от природы растворённого вещества и прямо пропорционально моляльной концентрации раствора.
![]()
Поскольку по мере кристаллизации растворителя из раствора концентрация последнего возрастает, растворы не имеют определённой температуры замерзания и кристаллизуются в некотором интервале температур.
Правило Вант-Гоффа — эмпирическое правило, позволяющее в первом приближении оценить влияние температуры на скорость химической реакции в небольшом температурном интервале (обычно от 0 °C до 100 °C). Я. Х. Вант-Гофф на основании множества экспериментов сформулировал следующее правило:
Уравнение, которое описывает это правило, следующее:
![]()
где
![]() —
скорость реакции при температуре
—
скорость реакции при температуре
![]() ,
,
![]() —
скорость реакции при температуре
—
скорость реакции при температуре
![]() ,
,
![]() —
температурный коэффициент реакции
(если он равен 2, например, то скорость
реакции будет увеличиваться в 2 раза
при повышении температуры на 10 градусов).
—
температурный коэффициент реакции
(если он равен 2, например, то скорость
реакции будет увеличиваться в 2 раза
при повышении температуры на 10 градусов).
Следует помнить, что правило Вант-Гоффа применимо только для реакций с энергией активации 60-120 кДж/моль в температурном диапазоне 10-400oC. Правилу Вант-Гоффа также не подчиняются реакции, в которых принимают участие громоздкие молекулы, например, белки в биологических системах. Температурную зависимость скорости реакции более корректно описывает уравнение Аррениуса.
Из уравнения Вант-Гоффа температурный коэффициент вычисляется по формуле:
![]()
Количественные характеристики растворов слабых электролитов. Закон Освальда.
Причину отклонения от законов Вант-Гоффа и Рауля впервые установил в 1887 г шведский ученый Сванте Аррениус, предложив теорию электролитической диссоциации, которая основывается на двух постулатах:
Вещества, растворы которых являются электролитами (т.е. проводят электрический ток), при растворении распадаются на частицы (ионы), которые образуются в результате диссоциации растворенного вещества. Число частиц при этом увеличивается. Ионы, заряженные положительно получили название катионы, т.к. под действием электрического поля движутся к катоду. Ионы заряженные отрицательно – анионы, т.к. под действием электрического поля движутся к аноду. К электролитам относятся соли, кислоты и основания.
Al(NO3)3 Al ³+ + NO3ֿ
Электролиты диссоциируют не полностью. Способность вещества к диссоциации характеризуется значением степени электролитической диссоциации - . Степенью электролитической диссоциации называется отношение количества вещества электролита, распавшегося на ионы, к общему количеству растворенного электролита.
= nионизированное / Nрастворенное
n-количество молекул распавшихся на ионы
N-общее количество молекул в растворе
- степень электролитической диссоциации
Значение может изменяться от 0 до 1, часто выражается в процентах (от 0 до 100%). Степень диссоциации показывает, какая часть растворенного количества электролита при данных условиях находится в растворе в виде гидратированных ионов.
Причины электролитической диссоциации обусловлены:
характером химических связей в соединениях (к электролитам относятся вещества с ионной или ковалентной сильнополярной связью)
характером растворителя: молекула воды полярна, т.е. является диполем
Таким образом, электролитической диссоциацией называют процесс распада ионных или полярных соединений на ионы под действием полярных молекул растворителя.
Механизм электролитической диссоциации.
Теорию Аррциуса значительно развили русские ученые И.А.Каблуков и В.А.Кистяковский, они доказали, что при растворении электролита происходит химическое взаимодействие растворенного вещества с водой, которое приводит к образованию гидратов, а затем они диссоциируют на ионы, т.е. в растворе находятся гидратированные ионы.
Легче всего диссоциация вещества с ионной связью. Последовательность процессов происходящих при диссоциации веществ с ионной связью (солей, щелочей) будет такой:
ориентация молекул диполей воды около ионов кристалла
гидратация (взаимодействие) молекул воды с ионами поверхностного слоя кристалла
диссоциация (распад) кристалла электролита на гидратированные ионы.
С учетом гидратации ионов уравнение диссоциации выглядит так:
NaCl + X H2O Na + n •H2O + Cl - n• H2O
Так как состав гидратированных ионов не всегда постоянен, уравнение записывают сокращенно:
NaCl Na+ + Cl-
Аналогично происходит и процесс диссоциации веществ с полярной связью, последовательность происходящих процессов следующая:
ориентация молекул воды вокруг полюсов молекулы электролита
гидратация (взаимодействие) молекулы воды с молекулами электролита
ионизация молекул электролита (превращение ковалентной полярной связи в ионную)
диссоциация (распад) молекул электролита на гидратированные ионы.
HCl + H2O H3O++ Cl-
HCl H+ + Cl-
В процессе диссоциации ион водорода в свободном виде не встречается, только в виде иона гидроксония H3O+.