

4 Конформационный анализ и исследование переходного состояния молекулы
Многие молекулы могут иметь несколько пространственных структур (конформаций), отличающихся взаимным положением атомных группировок, причем эти конформации, как правило, отличаются по энергии. Переход от одной конформации к другой связан с преодолением некоторого энергетического барьера. Раздел химии, занимающийся исследованием конформационных переходов, называется конформационным анализом. Подобный анализ весьма важен, поскольку различные конформации молекул могут отличаться реакционной способностью. Кроме того, термодинамическая и кинетическая гибкость полимерных цепей объясняется именно различными конформационными состояниями цепей макромолекул? отличающихся по величине потенциальной энергии.
Основное положение теории активированного комплекса заключается в том, что превращение исходных частиц (молекул, атомов, радикалов, ионов) в конечные продукты реакции протекает через переходное состояние (активированный комплекс)? когда в реакционной системе в реагентах исчезают отдельные связи, а в продуктах возникают новые.
Элементарный акт химической реакции сопровождается разрывом определенной связи (связей) в реагентах и образованием новых в продуктах, вследствие изменений расстояний между ядрами и перераспределения электронной плотности в системе под действием внешней энергии. Направление химической реакции определятся качеством (квантами энергии) и количеством энергии, перераспределяемой между реагентами, продуктами и внешней средой. При этом относительное расположение ядер меняется непрерывно, меняется при этом и распределение электронной плотности в реакционной системе. Для осуществления элементарного химического акта реагирующие молекулы должны преодолеть некоторый энергетический барьер, высота которого определяется природой трансформируемых частиц. Элементарный химический акт описывается движением точки по поверхности, характеризующей изменение потенциальной энергии по линии минимальных энергетических затрат, которую называют путем реакции или координатой реакции. Если получить зависимость изменения потенциальной энергии реакционной системы от координаты реакции, то можно найти минимальное значение энергии переходного состояния, которая определяет вершину энергетического барьера. Разность между энергией основного состояния активированного комплекса и энергией исходных реагирующих веществ дает истинную энергию активации реакции. Селективность перехода возбужденных частиц (активированного комплекса) в продукты реакции будет определяться качеством (квантами энергии) отдаваемой этим комплексом реакционной системе.

Рисунок 7 – Синильная кислота
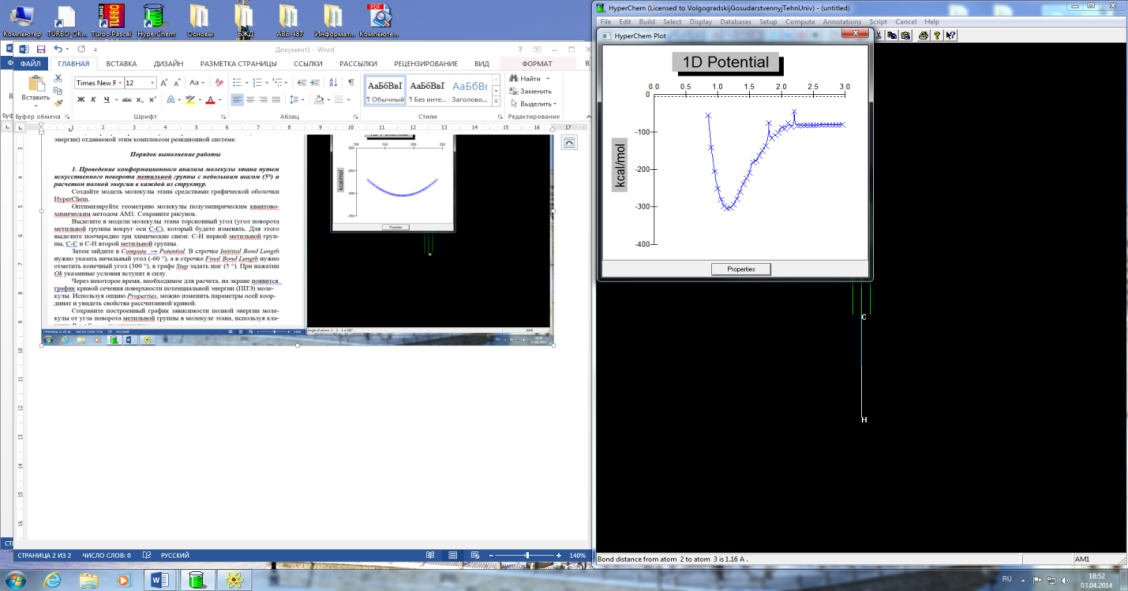
Рисунок 8 – график кривой сечения поверхности потенциальной энергии синильной кислоты.
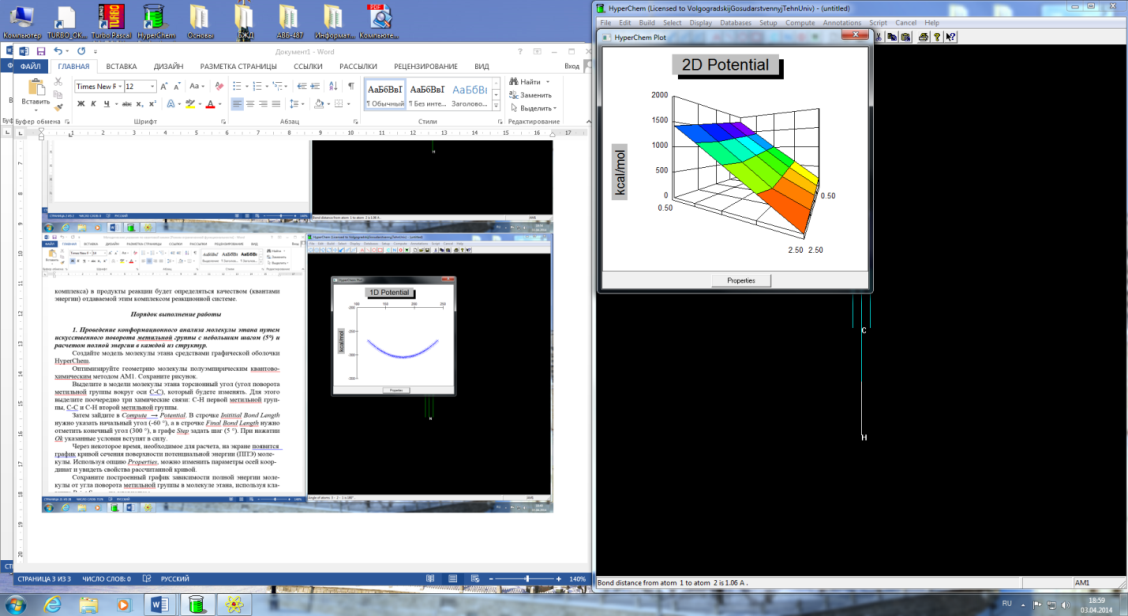
Рисунок 9 – График полной энергии синильной кислоты.
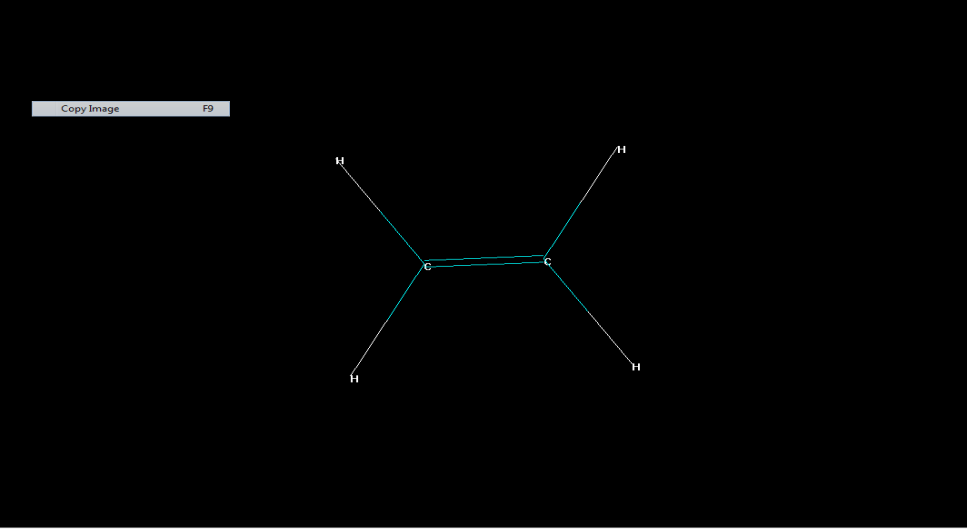
Рисунок 10 – Молекула этана.
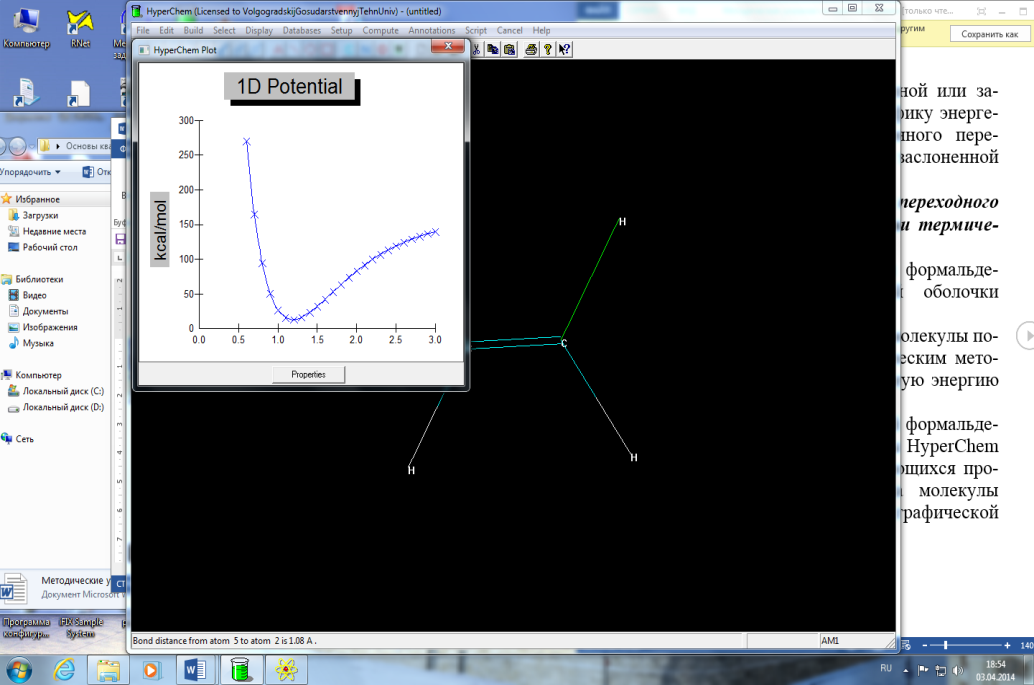
Рисунок12-График кривой сечения поверхности потенциальной энергии молекулы этана.

Рисунок 13 – График полной энергии молекулы этана
Определение структуры переходного состояния и энергии активации термического распада молекулы.
Рисунок 14- Молекулы муравьиного альдегида углекислого газа и водорода.
HyperChem log start -- Wed May 28 14:11:19 2014.
Geometry optimization, SemiEmpirical, molecule = (untitled).
AM1
PolakRibiere optimizer
Convergence limit = 0.0100000 Iteration limit = 50
Accelerate convergence = YES
Optimization algorithm = Polak-Ribiere
Criterion of RMS gradient = 0.1000 kcal/(A mol) Maximum cycles = 60
RHF Calculation:
Singlet state calculation
Number of electrons = 12
Number of Double Occupied Levels = 6
Charge on the System = 0
Total Orbitals = 10
Starting AM1 calculation with 10 orbitals
E=0.0000 kcal/mol Grad=0.000 Conv=NO(0 cycles 0 points) [Iter=1 Diff=3273.71050]
E=5064.3866 kcal/mol Grad=8969.884 Conv=NO(0 cycles 1 points) [Iter=1 Diff=47.08708]
E=2259.9987 kcal/mol Grad=4197.608 Conv=NO(0 cycles 2 points) [Iter=6 Diff=0.00478]
E=914.9524 kcal/mol Grad=2124.453 Conv=NO(0 cycles 3 points) [Iter=6 Diff=0.00502]
E=-121.2363 kcal/mol Grad=515.129 Conv=NO(0 cycles 4 points) [Iter=8 Diff=0.00124]
E=-195.4944 kcal/mol Grad=112.525 Conv=NO(0 cycles 5 points) [Iter=5 Diff=0.00753]
E=-276.4733 kcal/mol Grad=112.111 Conv=NO(0 cycles 6 points) [Iter=5 Diff=0.00448]
E=-248.2938 kcal/mol Grad=215.694 Conv=NO(1 cycles 7 points) [Iter=4 Diff=0.00592]
E=-290.8751 kcal/mol Grad=179.511 Conv=NO(1 cycles 8 points) [Iter=3 Diff=0.01437]
E=-318.1519 kcal/mol Grad=153.054 Conv=NO(1 cycles 9 points) [Iter=5 Diff=0.00064]
E=-334.0930 kcal/mol Grad=125.717 Conv=NO(1 cycles 10 points) [Iter=2 Diff=0.00440]
Geometry optimization, SemiEmpirical, molecule = (untitled).
AM1
PolakRibiere optimizer
Convergence limit = 0.0100000 Iteration limit = 50
Accelerate convergence = YES
Optimization algorithm = Polak-Ribiere
Criterion of RMS gradient = 0.1000 kcal/(A mol) Maximum cycles = 60
RHF Calculation:
Singlet state calculation
Number of electrons = 12
Number of Double Occupied Levels = 6
Charge on the System = 0
Total Orbitals = 10
Starting AM1 calculation with 10 orbitals
E=0.0000 kcal/mol Grad=0.000 Conv=NO(0 cycles 0 points) [Iter=1 Diff=2023.99913]
E=-248.2938 kcal/mol Grad=215.693 Conv=NO(0 cycles 1 points) [Iter=1 Diff=79.13621]
E=-248.2938 kcal/mol Grad=215.693 Conv=NO(0 cycles 1 points) [Iter=10 Diff=0.00740]
E=-162.2645 kcal/mol Grad=80.635 Conv=NO(0 cycles 2 points) [Iter=6 Diff=0.00185]
E=-303.8142 kcal/mol Grad=106.448 Conv=NO(1 cycles 3 points) [Iter=5 Diff=0.00051]
E=-336.3804 kcal/mol Grad=80.381 Conv=NO(1 cycles 4 points) [Iter=5 Diff=0.00058]
E=-345.1257 kcal/mol Grad=61.363 Conv=NO(2 cycles 6 points) [Iter=5 Diff=0.00665]
E=-291.9835 kcal/mol Grad=301.539 Conv=NO(2 cycles 7 points) [Iter=5 Diff=0.00283]
E=-352.1302 kcal/mol Grad=28.504 Conv=NO(3 cycles 8 points) [Iter=2 Diff=0.00510]
E=-356.0861 kcal/mol Grad=23.670 Conv=NO(3 cycles 9 points) [Iter=2 Diff=0.00509]
E=-359.2680 kcal/mol Grad=18.916 Conv=NO(3 cycles 10 points) [Iter=3 Diff=0.00270]
E=-363.0899 kcal/mol Grad=14.418 Conv=NO(3 cycles 11 points) [Iter=1 Diff=0.75471]
E=-363.0899 kcal/mol Grad=14.418 Conv=NO(3 cycles 11 points) [Iter=4 Diff=0.00219]
E=-356.7272 kcal/mol Grad=67.610 Conv=NO(4 cycles 14 points) [Iter=5 Diff=0.00058]
E=-365.8170 kcal/mol Grad=15.803 Conv=NO(5 cycles 15 points) [Iter=1 Diff=1.37372]
E=-359.9586 kcal/mol Grad=57.275 Conv=NO(5 cycles 16 points) [Iter=4 Diff=0.00322]
E=-366.1750 kcal/mol Grad=2.143 Conv=NO(7 cycles 23 points) [Iter=1 Diff=0.00108]
ENERGIES AND GRADIENT
Total Energy = -10967.5088290 (kcal/mol)
Total Energy = -17.477835678 (a.u.)
Binding Energy = -366.1826410 (kcal/mol)
Isolated Atomic Energy = -10601.3261880 (kcal/mol)
Electronic Energy = -19930.9771032 (kcal/mol)
Core-Core Interaction = 8963.4682741 (kcal/mol)
Heat of Formation = -31.5296410 (kcal/mol)
Gradient = 0.0371753 (kcal/mol/Ang)
MOLECULAR POINT GROUP
C2V
EIGENVALUES(eV)
Symmetry: 1 A1 2 A1 1 B2 3 A1 1 B1
Eigenvalue: -39.064316 -25.061728 -17.138297 -16.260277 -14.544736
Symmetry: 2 B2 2 B1 4 A1 3 B2 5 A1
Eigenvalue: -10.782875 0.793200 3.166694 3.996373 6.066060
ATOMIC ORBITAL ELECTRON POPULATIONS
AO: 1 S C 1 Px C 1 Py C 1 Pz C 2 S O
1.281447 0.957209 0.922478 0.700426 1.909529
AO: 2 Px O 2 Py O 2 Pz O 3 S H 4 S H
1.748514 1.318238 1.299574 0.931293 0.931293
NET CHARGES AND COORDINATES
Atom Z Charge Coordinates(Angstrom) Mass
x y z
1 6 0.138440 -0.10111 0.38253 0.00000 12.01100
2 8 -0.275854 -0.67058 -0.70465 -0.00000 15.99900
3 1 0.068707 -0.65832 1.34311 0.00000 1.00800
4 1 0.068707 1.00582 0.47142 0.00000 1.00800
ATOMIC GRADIENTS
Atom Z Gradients(kcal/mol/Angstrom)
x y z
1 6 -0.05091 -0.09693 0.00000
2 8 0.02068 0.03946 -0.00000
3 1 0.02915 0.02135 -0.00000
4 1 0.00108 0.03612 0.00000
Dipole (Debyes) x y z Total
Point-Chg. 0.936 1.787 0.000 2.017
sp Hybrid 0.139 0.266 0.000 0.300
pd Hybrid 0.000 0.000 0.000 0.000
Sum 1.075 2.052 0.000 2.317
Transition State Search: Eigenvector Following, SemiEmpirical, molecule = (untitled).
AM1
Convergence limit = 0.0100000 Iteration limit = 50
Accelerate convergence = YES
RHF Calculation:
Singlet state calculation
Number of electrons = 12
Number of Double Occupied Levels = 6
Charge on the System = 0
Total Orbitals = 10
Starting AM1 calculation with 10 orbitals
Computing the Hessian is required.
Computing the Hessian using Cartesian coordinates.
Iteration = 1 Difference = 1884.15592
Iteration = 2 Difference = 49.72128
Iteration = 3 Difference = 2.07549
Iteration = 4 Difference = 0.05117
Iteration = 5 Difference = 0.06324
Iteration = 6 Difference = 0.00138
Computing the initial Hessian: done 20%.
Computing the initial Hessian: done 50%.
Computing the initial Hessian: done 70%.
Computing the initial Hessian: done 100%.
ENERGIES AND GRADIENT
Total Energy = -10966.2256533 (kcal/mol)
Total Energy = -17.475790808 (a.u.)
Binding Energy = -364.8994653 (kcal/mol)
Isolated Atomic Energy = -10601.3261880 (kcal/mol)
Electronic Energy = -20031.3226545 (kcal/mol)
Core-Core Interaction = 9065.0970012 (kcal/mol)
Heat of Formation = -30.2464653 (kcal/mol)
Gradient = 15.5092916 (kcal/mol/Ang)
MOLECULAR POINT GROUP
C2V
EIGENVALUES(eV)
Symmetry: 1 A1 2 A1 1 B2 3 A1 1 B1
Eigenvalue: -39.342077 -25.231336 -17.432142 -16.216731 -14.629620
Symmetry: 2 B2 2 B1 4 A1 3 B2 5 A1
Eigenvalue: -10.829941 0.829538 3.332886 4.196891 6.061152
ATOMIC ORBITAL ELECTRON POPULATIONS
AO: 1 S C 1 Px C 1 Py C 1 Pz C 2 S O
1.269676 0.968072 0.911316 0.707462 1.907594
AO: 2 Px O 2 Py O 2 Pz O 3 S H 4 S H
1.915965 1.164187 1.292539 0.931594 0.931594
NET CHARGES AND COORDINATES
Atom Z Charge Coordinates(Angstrom) Mass
x y z
1 6 0.143473 -0.51447 -1.41046 -0.00000 12.01100
2 8 -0.280286 -0.51447 -0.19046 -0.00000 15.99900
3 1 0.068406 0.42084 -1.95046 -0.00000 1.00800
4 1 0.068406 -1.44978 -1.95046 -0.00000 1.00800
ATOMIC GRADIENTS
Atom Z Gradients(kcal/mol/Angstrom)
x y z
1 6 -0.00005 -7.04928 0.01225
2 8 -0.00002 -34.76636 0.01210
3 1 -19.41352 20.90782 0.08397
4 1 19.41359 20.90782 -0.10832
Dipole (Debyes) x y z Total
Point-Chg. 0.000 -1.997 -0.000 1.997
sp Hybrid -0.000 -0.297 0.000 0.297
pd Hybrid 0.000 0.000 0.000 0.000
Sum 0.000 -2.294 0.000 2.294
Transition State Search: Eigenvector Following, SemiEmpirical, molecule = (untitled).
AM1
Convergence limit = 0.0100000 Iteration limit = 50
Accelerate convergence = YES
RHF Calculation:
Singlet state calculation
Number of electrons = 12
Number of Double Occupied Levels = 6
Charge on the System = 0
Total Orbitals = 10
Starting AM1 calculation with 10 orbitals
Using the original Hessian.
The eigenvector-follow algorithm is used for transition state search with Cartesian coordinates.
The input mode is 1.
E=-364.8997 kcal/mol Grad=15.509 NumOfNegEigenvalues=0 Conv=NO(1 cycles 1 points) [Iter=1 Diff=0.20828]
E=-353.7406 kcal/mol Grad=23.028 NumOfNegEigenvalues=0 Conv=NO(3 cycles 3 points) [Iter=4 Diff=0.00886]
E=-319.1363 kcal/mol Grad=42.780 NumOfNegEigenvalues=0 Conv=NO(4 cycles 4 points) [Iter=5 Diff=0.00046]
E=-268.5120 kcal/mol Grad=53.604 NumOfNegEigenvalues=0 Conv=NO(5 cycles 5 points) [Iter=1 Diff=2.71160]
E=-215.3580 kcal/mol Grad=51.863 NumOfNegEigenvalues=1 Conv=NO(6 cycles 6 points) [Iter=5 Diff=0.00627]
E=-173.5125 kcal/mol Grad=38.968 NumOfNegEigenvalues=1 Conv=NO(7 cycles 7 points) [Iter=1 Diff=4.61699]
E=-148.2129 kcal/mol Grad=21.671 NumOfNegEigenvalues=1 Conv=NO(8 cycles 8 points) [Iter=4 Diff=0.22627]
E=-137.4411 kcal/mol Grad=13.599 NumOfNegEigenvalues=1 Conv=NO(9 cycles 9 points) [Iter=5 Diff=0.
E=-130.7338 kcal/mol Grad=8.577 NumOfNegEigenvalues=1 Conv=NO(10 cycles 10 points) [Iter=5 Diff=0.00293]
E=-125.3736 kcal/mol Grad=6.096 NumOfNegEigenvalues=1 Conv=NO(11 cycles 11 points) [Iter=5 Diff=0.01215]
E=-121.0339 kcal/mol Grad=4.257 NumOfNegEigenvalues=1 Conv=NO(12 cycles 12 points) [Iter=3 Diff=0.02192]
E=-117.6962 kcal/mol Grad=2.658 NumOfNegEigenvalues=1 Conv=NO(16 cycles 16 points) [Iter=50 Diff=0.12982]
E=6.0474 kcal/mol Grad=7.605 NumOfNegEigenvalues=1 Conv=NO(16 cycles 24 points) [Iter=13 Diff=0.06610]
HyperChem log stop -- Wed May 28 14:17:53 2014.