

Министерство образования и науки Российской Федерации и
Волгоградский государственный технический университет
Кафедра
семестровое задание № 1
Тема: Основы квантовохимического анализа
Основы квантовохимического анализа .
.
Выполнил
Студент гр. ТВБ-385
Конюкова М.П.
Проверил
асс.Климов В.В.
Волгоград 2014
Введение
1 Знакомство с программным комплексом HYPERCHEM.Построение и редактирование химических структур
2 Оптимизация геометрии молекул, определение распределения эффектив- ных атомных зарядов и пространственной формы граничных орбиталей
3 Конформационный анализ и исследование переходного состояния молекулы
4 Конформационный анализ и исследование переходного состояния молекулы
5 Исследование колебательного спектра молекул
Выводы
Список литературы
Введение
Классическая теория химического строения позволяет трактовать, какова может быть структура химического соединения, каковы особенности этой структуры и свойств рассматриваемого соединения сравнительно с другими соединениями, каков набор химических и физико-химических свойств должен быть присущ этому соединению. Квантовая теория оперирует более детальной информацией о строении вещества, что позволяет ей объяснять и предсказывать многие свойства химических соединений, в том числе такие, которые подчас неподвластны классической теории, например свойства возбужденных состояний.
Квантовая химия – это раздел теоретической химии, в котором строение и свойства соединений рассматриваются на основе представлений квантовой механики и экспериментально установленных закономерностей. Квантовая химия использует математический аппарат и методы квантовой механики для описания и расчета свойств химических соединений, начиная с атомов и простейших молекул и кончая высокомолекулярными соединениями и кристаллическими системами.
На сегодняшний день методы квантовой химии и молекулярной динамики получили широкое распространение в численном моделировании электронной и атомной структур сложных систем молекулярных, кристаллических и переходных (нано) размеров. Это связано с технологическим развитием соответствующего математического обеспечения. В настоящее время создано достаточно много современных вычислительных комплексов, реализующих методы квантовой химии и молекулярной динамики, однако, причем для широкого круга пользователей наиболее доступно использование этих методов обеспечивается квантовохимической и молекулярно-динамической программой HyperChem.
1 Знакомство с программным комплексом hyperchem.Построение и редактирование химических структур
Программный комплекс HyperChem позволяет проводить следующий типы вычислений:
вычисление молекулярной энергии и других характеристик молекулярной системы с фиксированной геометрией;
оптимизация геометрии путем минимизации полной энергии системы;
вычисление частот нормальных колебаний структур с оптимизированной геометрией;
изображение колебательного спектра, а также колебательных движений, связанных с конкретными колебательными состояниями;
поиск переходного состояния, то есть, поиск структур, соответствующих переходному состоянию химической реакции;
моделирование молекулярной динамики: вычисление классических траекторий для молекулярных систем и другие вычисления.
Программа HyperChem может выполнять расчеты энергии систем и их равновесной геометрии методом молекулярной механики с использованием четырех модельных потенциалов (MM+, AMBER, BIO+ и OPLS), девятью полуэмпирическими квантово-химическими методами (Расширенный метод Хюккеля, CNDO, INDO, MINDO3, MNDO, AM1, PM3, ZINDO/1 и ZINDO/S), или неэмпирическим (ab initio) методом квантовой химии в различных базисах.
Расчет энергии фенола методом молекулярной механики ММ+
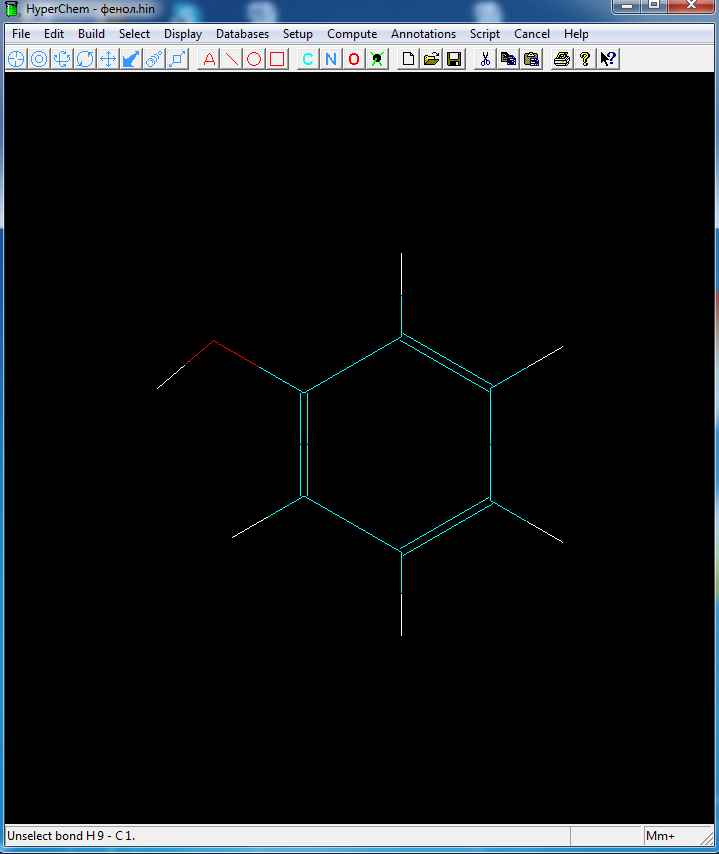
Рисунок 1- фенол
forcefield mm+
sys 0 0 1
view 40 0.36488 55 15 1 0 0 0 1 0 0 0 1 0.1987 -0.43578 -55
seed -1110
mol 1
atom 1 - C ** - 0 -0.6221451 1.754659 0.2173739 3 2 d 6 s 8 s
atom 2 - C ** - 0 -1.498884 0.764467 -0.02745624 3 1 d 3 s 9 s
atom 3 - C ** - 0 -1.020901 -0.6134776 -0.2143136 3 2 s 4 d 10 s
atom 4 - C ** - 0 0.2961497 -0.8882096 -0.141486 3 3 d 5 s 7 s
atom 5 - C ** - 0 1.252352 0.1978559 0.1281533 3 4 s 6 d 11 s
atom 6 - C ** - 0 0.8150121 1.458083 0.3000144 3 5 d 1 s 12 s
atom 7 - O ** - 0 0.766612 -2.147846 -0.3107609 2 4 s 13 s
atom 8 - H ** - 0 -0.9622551 2.740256 0.3500846 1 1 s
atom 9 - H ** - 0 -2.527159 0.9740889 -0.08741576 1 2 s
atom 10 - H ** - 0 -1.71431 -1.380659 -0.4046163 1 3 s
atom 11 - H ** - 0 2.280781 -0.01237505 0.1862238 1 5 s
atom 12 - H ** - 0 1.499561 2.231943 0.4930395 1 6 s
atom 13 - H ** - 0 0.1910408 -2.870297 -0.4888407 1 7 s
endmol 1
HyperChem log start -- Mon May 19 09:47:16 2014.
Geometry optimization, SemiEmpirical, molecule = C:\Users\родители\Desktop\фенол.hin.
PolakRibiere optimizer
Convergence limit = 0.0100000 Iteration limit = 50
Accelerate convergence = YES
Optimization algorithm = Polak-Ribiere
Criterion of RMS gradient = 0.1000 kcal/(A mol) Maximum cycles = 195
RHF Calculation:
Singlet state calculation
Number of electrons = 36
Number of Double Occupied Levels = 18
Charge on the System = 0
Total Orbitals = 34
E=-1418.8161 kcal/mol Grad=10.434 Conv=NO(3 cycles 9 points) [Iter=3 Diff=0.00769]
E=-1419.9177 kcal/mol Grad=0.179 Conv=NO(15 cycles 34 points) [Iter=1 Diff=0.00043]
E=-1419.9193 kcal/mol Grad=0.094 Conv=YES(18 cycles 40 points) [Iter=1 Diff=0.00000]
ENERGIES AND GRADIENT
Total Energy = -27003.1469454 (kcal/mol)
Total Energy = -43.032248477 (a.u.)
Binding Energy = -1419.9192534 (kcal/mol)
Isolated Atomic Energy = -25583.2276920 (kcal/mol)
Electronic Energy = -102767.4934862 (kcal/mol)
Core-Core Interaction = 75764.3465408 (kcal/mol)
Heat of Formation = -22.4082534 (kcal/mol)
Gradient = 0.0938265 (kcal/mol/Ang)
MOLECULAR POINT GROUP
CS
AM1
EIGENVALUES(eV)
Symmetry: 1 A' 2 A' 3 A' 4 A' 5 A'
Eigenvalue: -40.144963 -37.383381 -31.620205 -30.258145 -23.684800
Symmetry: 6 A' 7 A' 8 A' 9 A' 1 A"
Eigenvalue: -23.003528 -18.999511 -17.424423 -16.545861 -15.048749
Symmetry: 10 A' 11 A' 12 A' 2 A" 13 A'
Eigenvalue: -15.034117 -14.406476 -14.271576 -12.644716 -12.505277
Symmetry: 14 A' 3 A" 4 A" 5 A" 6 A"
Eigenvalue: -11.940192 -9.850798 -9.115071 0.397795 0.508548
Symmetry: 7 A" 15 A' 16 A' 17 A' 18 A'
Eigenvalue: 2.839988 3.025781 3.801153 3.835648 4.082076
Symmetry: 19 A' 20 A' 21 A' 22 A' 23 A'
Eigenvalue: 4.306876 4.423635 4.720782 5.037207 5.220481
Symmetry: 24 A' 25 A' 26 A' 27 A'
Eigenvalue: 5.419090 5.612412 5.836334 6.320537
ATOMIC ORBITAL ELECTRON POPULATIONS
AO: 1 S C 1 Px C 1 Py C 1 Pz C 2 S C
1.217088 0.927757 0.985179 1.035560 1.215534
AO: 2 Px C 2 Py C 2 Pz C 3 S C 3 Px C
0.988330 0.916897 0.970844 1.218175 0.929738
AO: 3 Py C 3 Pz C 4 S C 4 Px C 4 Py C
0.976422 1.089054 1.200156 0.914230 0.850711
AO: 4 Pz C 5 S C 5 Px C 5 Py C 5 Pz C
0.956982 1.214013 0.988151 0.908416 1.046077
AO: 6 S C 6 Px C 6 Py C 6 Pz C 7 S O
1.216861 0.950940 0.955419 0.973789 1.862648
AO: 7 Px O 7 Py O 7 Pz O 8 S H 9 S H
1.338590 1.165262 1.886127 0.866736 0.867839
AO: 10 S H 11 S H 12 S H 13 S H
0.867355 0.850352 0.866082 0.782686