

1.Титриметрический анализ. Основные понятия (аликвота, титрант, точка эквивалентности, индикатор, кривая тирования). Требования, предъявляемые к реакциям в титриметрии. Реактивы, применяемые в титриметрии. Стандартные вещества, титранты.
- метод количественного анализа, основанный на измерении объёма раствора с точно известной концентрацией реактива, требующегося для реакции с данным количеством определяемого вещества.Аликвота-точно измеренная кратная часть образца (объём раствора), взятая для анализа, которая сохраняет свойства основного образца.Титрант или рабочий раствор - это раствор, с помощью которого проводят титрование. Точка эквивалентности момент титрования, когда количество добавленного титранта химически эквивалентно количеству титруемого вещества. ТЭ еще можно назвать стехиометрической точкой, теоретической конечной точкой.Индикатор- вещество, которое изменяет свою окраску в ТЭ, характеризуется малой концентрацией и интервалом перехода. Кривая титрования-показывает графическую зависимость логарифма концентрации участника реакции, протекающей при титровании, или к-то св-ва р-ра от объема добавленного титранта(или от степени оттитрованности).Н-р, для реакции кисл-осн взаимод-я кривые титр. Строят в координатах рН-объем титранта.
Требования к реакциям в титриметрии: 1.Взаимодействие титранта с определяемым веществом должно проходить в точном соответствии со стехиометрическим уравнением реакции, и титрант должен расходоваться только на реакцию с определяемым веществом. В то же время определяемое вещество должно реагировать только с титрантом и не взаимодействовать, например, с кислородом атмосферы, как это в принципе может быть при титровании восстановителей.
2. Реакция титрования должна протекать количественно, т. е. константа равновесия реакции титрования должна быть достаточно велика.
Взаимодействие определяемого вещества с титрантом должно проходить с большой скоростью.
Должен существовать способ, позволяющий определять окончание титрования.
5. Раствор титранта должен быть стандартизирован. Реактивы: По свойствам веществ и способу их приготовления титранты бывают двух типов: стандартные, с приготовленным титром, стандартизированные или с установленным титром. Стандартные растворы или с приготовленным титром называют первичный стандартный раствор. Он готовится растворением точного количества чистого химического вещества в определенном объеме растворителя. К первичным стандартным веществам относятся: Na2CO3, Na2B4O7*10H2O, Na2SO4, CaCO3, CaCI2, MgSO4, MgCI2, H2C2O4*2H2O, Na2C2O4, K2Cr2O7, бикарбонат натрия, бромат калия, иодат калия и другие.
Первый тип титрантов (с приготовленным титром) применяют в титриметрии для количественных определений тех или иных веществ и для установки титров второго типа - вторичных стандартных растворов.
Вторичный стандартный раствор - это растворы таких веществ, концентрация которых устанавливается (стандартизируется) по концентрации первичных стандартных растворов или готовится по известной массе вторичного стандартного вещества.
Ко второму типу титрантов относят растворы таких веществ, которые не удовлетворяют требованиям, предъявляемые к первичным стандартным веществам. К ним относятся: щелочи, растворы кислот HCI, H2SO4, HNO3, CH3COOH, KMnO4, AgNO3, Na2S2O3 и другие.
2. Типовые расчеты в титриметрии. Способы выражения концентраций в титриметрии (молярная концентрация, молярная концентрация эквивалента, титр, поправочный коэффициент. Расчет массы стандартного образца для приготовления титранта, расчет концентрации титранта
Молярная концентрация с(А) — количество растворенного вещества А в молях, содержащееся в одном литре раствора: моль/л. с(А) = n(А)/V(А) = m(А)/M/(А)V(А),где п(А) — количество растворенного вещества А, моль; V(А) — объем раствора, л; т(А) — масса растворенного вещества А, г; М/(А) — молярная масса растворенного вещества А, г/моль.Молярная концентрация эквивалента с(1/zА), ,— количество растворенного вещества А в молях, соответствующее эквиваленту А, содержащееся в одном литре раствора: моль/л с(1/z А) = п(1/z А)/V(А) = т(А)/М(1/zА)V(А),где 1/z — фактор эквивалентности; рассчитывается для каждого вещества на основании стехиометрии реакции; п(1/zА) — количество вещества, эквиваленту А в растворе, моль; М(1/zА) — молярная масса эквивалента растворенного вещества А, г/моль. Титр Т(А) растворенного вещества А — это масса растворенного в-ва А, содержащаяся в одном мл раствора:измер в мл Т(А) = m(А)/V(А) = с(1/z А)М(1/z А)/1000.Титр раствора по определяемому веществу X, или титриметриче-ский фактор пересчета t(Т/Х), — масса титруемого вещества X, взаимодействующая с одним мл титранта Т: t(Т/Х) = Т(Т)М(1/zХ) /М(1/zТ) = с(1/zT) М(1/zХ)/1000 .Измеряется в г/мл.Поправочный коэффициент F (или К) — число, выражающее отношение действительной (практической) концентрации с(1/zА)пр вещества А в растворе к его заданной (теоретической) концентрации с(1/z А)теор: F = с(1/zА)пр/с(1/zА)теор. Расчет массы навески стандартного вещества. Массу навески т(А) стандартного вещества А, необходимую для получения раствора с заданной молярной концентрацией эквивалента с(1/zА), рассчитывают по формуле:m(А) = с(1/z А)М(1/z А)VА),где М(1/z А) — молярная масса эквивалента вещества А.Если задается молярная концентрация с(А), то масса навески вычисляется аналогично по формуле:т(А) = с(А)М(А)V(А),Где M/(А) — молярная масса вещества А. Массу навески обычно взвешивают на аналитических весах с ошибкой взвешивания ±0,0002 г. Расчет концентрации титранта Т при его стандартизации по стандартному раствору вещества А проводят следующим образом. Пусть при стандартизации протекает реакция Т + А = В.Согласно закону эквивалентов, эквивалентные количества веществ Т,А и В равны n (1/z Т) = n (1/z А) = n (1/z В), эквивалентное количество вещества равно произведению молярной концентрации эквивалента этого вещества на объем его раствора: с(1/z Т) = с(1/z А)V(А)/V(Т) = с(1/z В) V(В)/V(Т).
3. Классификация методов титриметрического анализа – кислотно – основное, окислительно – восстановительное, осадительное, комплексонометрическое. Виды титрования (прямое, обратное, косвенное). Методы установления точки титрования.
1) Кислотно-основное титрование (метод нейтрализации) — тит рование, основанное на реакции переноса протонов от одной реагирующей частицы к другой в растворе. Различают ацидиметрию и алкалиметрию.
Ацидиметрия (ацидиметрическое титрование) — определение веществ титрованием стандартным раствором кислоты.
Алкалиметрия (алкалиметрическое титрование) — определение веществ титрованием стандартным раствором сильного основания.
2) Окислительно-восстановительное титрование (редоксметрия) — титрование, сопровождаемое переходом одного или большего числа
электронов от иона-донора или молекулы (восстановителя) к акцептору окислителю).
3) Осадителъное титрование — такое титрование, когда титруемое в-во при взаимодействии с титрантом выделяется из раствора в виде осадка
4) Комплексиметрическое титрование — титрование вещества раствором[ такого соединения, которое образует с титруемым веществом слабодиссоциирующий растворимый комплекс.
Разновидностью комплексиметрического титрования является комплексонометрическое титрование (комплексонометрия) — такое титрование когда титруемое вещество при взаимодействии с титрантом — раствором комплексонов — образует комплексонаты металлов.
Прямое титрование — это такое титрование, когда определяемое вещество непосредственно титруется стандартным раствором титранта или наоборот. Обратное титрование (титрование по остатку) — титрование не прореагировавшего вещества, которое прибавлено в избытке к анализи- руемому раствору в виде стандартного раствора. Косвенное титрование (заместительное титрование) — титрование, при котором определяемое вещество не реагирует с титрантом непосредственно, а определяется косвенно в результате использования сте-хиометрически протекающей реакции с образованием другого вещества, реагирующего с титрантом. Методы установления конечной точки титрованияСуществуют две группы методов фиксирования КТТ: визуальные и инструментальные.
Визуальные методы. За ходом реакции следят визуально, наблюдая изменение окраски (или другого свойства) специально внесенного индикатора| при нейтрализации, окислении-восстановлении, осаждении или комплексообразовании. КТТ устанавливают по резкому изменению видимого свойства системы в присутствии индикатора или без него: появление, изменение, исчезновение окраски, образование или растворение осадка.В индикаторных визуальных методах в титруемый раствор вносят индикатор. В безиндикаторных визуальных методах используют окраску титранта или титруемого вещества. КТТ определяют по появлению окраски титранта или по исчезновению окраски титруемого вещества.
Инструментальные методы. КТТ устанавливают по изменению физико-химических свойств раствора — флуоресценции, оптической плотности, потенциала, удельной электропроводности, силы тока, радиоактивности и др. Изменение физико-химических свойств фиксируют на различных приборах.
4. Кислотно – основное титрование. Основные реакции и титранты метода. Типы кислотно – основного титрования (алкалиметрия и ацидиметрия). Индикаторы, требования, предъявляемые к ним. Ионная, хромофорная, ионно- хромофорная теории индикаторов кислотно – основного титрования.
КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ— это метод определения кислот, оснований, солей, основанный на реакции взаимодействия между прото-литами — кислотой НА и основанием В:НА + В = А" + НВ+ В водных растворах — это реакция нейтрализации Н30++0Н=2Н20 поэтому метод кислотно-основного титрования также называют методом нейтрализации. Титрантами метода являются растворы сильных кислот и оснований: НС1, Н2S04, NаОН, КОН. Эти вещества не соответствуют требованиям, предъявляемым к стандартным веществам, поэтому концентрацию тит-рантов устанавливают стандартизацией их растворов. В качестве первичных стандартов чаще всего используют буру Nа2В407 • 10Н2О, безводный карбонат натрия Na2С03, дигидрат щавелевой кислоты Н2С204 • 2Н20 и некоторые другие. Ацидиметрическое титрование {ацидиметрия) — метод определения сильных и слабых оснований, солей слабых кислот, основных солей и других соединений, обладающих основными свойствами, путем титрования стандартным раствором сильной кислоты. Алкалиметрическое титрование {алкалиметрия) — метод определения сильных и слабых кислот, кислых солей, солей слабых оснований путем титрования стандартным раствором сильного основания.Индикатор — это вещество, которое проявляет видимое изменение в точке эквивалентности или вблизи ее.
Кислотно-основной индикатор сам является кислотой или основанием и при кислотно-основном титровании изменяет свою окраску в ТЭ или
вблизи ее. (Метиловый оранжевый рТ=4 Интервал перехода рН и окраска индикатора 3,1–4,4 Красная – оранжево-желтая; Фенолфталеин рТ=9,0 8,2–10 Бесцветная – фиолетовая).
Требования к индикаторам:1) окраска д.б. интенсивной,отлич-ся в кислой и щелочной среде 2) изменение окраски д.б. четким в узком интервале рН р-ра 3) индикатор д.б. чувствительным 4) инд-р д.б. стабильным, не разлагаться на воздухе, в р-ре. Теории индикаторов:
1)ионная (теория Оствальда)-индикаторы это слабые кислоты или основания, кот.ионизируют в водных растворах
HInd↔H+ +Ind-. Недостатки:1)она лишь констатирует различия окраски в кислой и щел. Ср.,но не объясняет природы окраски2)ионная р-я протекает мгновенно,а окраску индикатор меняет лишь со временем
2)Хромофорная-наличие окраски объясн-ся появлением хромофорных групп.Инд-ры в р-ре присут-ют в виде таутомерных форм.Недостатки:не объяс-ет почему происх-ят таутомерные превращения при изм-ии рН.
3)ионно-хромофорная- кислотно-основные индикаторы представляют собой слабые кислоты и основания, причем нейтральная молекула индикатора и ее ионизированная форма содержат разные хромофорные группы. Молекулы индикатора в водном растворе способны либо отдавать ионы водорода (индикатор — слабая кислота), либо принимать их (индикатор — слабое основание), подвергаясь при этом таутомерным превращениям.
Индикатор |
Изменение окраски |
Тимоловый синий |
красная — желтая |
Метиловый желтый |
то же |
Метиловый красный |
красная — желтая |
Феноловый красный |
желтая — красная |
Тимоловый синий |
тоже |
Фенолфталеин |
бесцветная — красная |
|
|
РЕАКЦИЯ(см тетрадь тема кисл-осн титрование)
5. Кривые кислотно – основного титрования. Расчет, построение и анализ типичных кривых титрования сильной кислоты щелочью и сильного и слабого основания кислотой. Выбор индикаторов по кривой титрования. Титрование полипротонных кислот. Ошибки кислотно – основного титрования, их расчет и устранение.
Кривые кислотно-основного титрования графически отображают зависимость изменения рН титруемого раствора от объема прибавленного титранта или от степени оттитрованности f= V(T)/V, где V(Т) и V— со ответственно объем прибавленного титранта в данный момент и в ТЭ.Чаще всего (хотя не всегда) при построении кривых кислотно- основного титрования вдоль оси абсцисс откладывают объем прибавлен- ного титранта или степень оттитрованности, а вдоль оси ординат — зна чения рН титруемого раствора..
Расчет, построение и анализ кривых титрования. Для построе ния кривой кислотно-основного титрования рассчитывают значения рН титруемого раствора в различные моменты титрования, т.е. в разных точках титрования: для исходного раствора, для растворов до ТЭ, в ТЭ и после ТЭ.
После начала титрования и до ТЭ значение рН раствора определяется как рН = -18с(Х)
Расчет рН в точке квивалентности. При титровании сильной кислоты сильным основанием среда в ТЭ — нейтральная, рН = 7.
Расчет рН после ТЭ. определяется концентрацией с(Т) щелочи, прибавлен-ной сверх стехиометрического количества. Учитывая, что рН + рОН = 14 можно написать:рН=14-рОН
П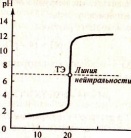 о
формулам рассчитывают значения рН
раствора в различные моменты титрования,
и по рассчитанным данным строят кривую
титрования в координатах рН—V(Т).
о
формулам рассчитывают значения рН
раствора в различные моменты титрования,
и по рассчитанным данным строят кривую
титрования в координатах рН—V(Т).
Рассчитанная кривая титрования 20 мл 0,1000 моль/л раствора НС1 0,1000 моль/л раствором №ОН
Для определения КТТ в данном случае можно использовать такие индикаторы кислотно-основного титрования, как метиловый оранжевый (рТ = 4), метиловый красный (рТ = 5,5), бромтимоловый синий (рТ = 7,0), фенолфталеин (рТ = 9) и др., для которых величина рТ лежит в интервале от 3 до 11. Чаще всего применяют метиловый оранжевый и фенолфталеин как наиболее доступные индикаторы кислотно-основного титрования. Обычно стремятся выбрать индикатор так, чтобы, при прочих равных условиях, значение рТ индикатора было бы как можно ближе к величине рН раствора в ТЭ, так как при этом уменьшается ошибка титрования.
Титрование сильного основания сильной кислотой. При титровании сильного основания сильной кислотой, например раствора гидроксида Натрия раствором хлороводородной кислоты, протекают процессы, аналогичные рассмотренным в предыдущем разделе, но только в обратном Направлении: по мере прибавления титранта значение рН раствора не увеличивается, а уменьшается.Для исходного раствора сильного основания и титруемого раствора величина рН до ТЭ определяется концентрацией щелочи в растворе. В ТЭ раствор — нейтральный, рН = 7.После ТЭ величина рН раствора обусловлена присутствием избы' точного титранта — сильной кислоты
Титрование поликислотных оснований. Растворы поликислотных оснований титруются раствором сильной кислоты последовательно, ступенчато. При приемлемом уровне титрования скачки на кривой титрования разделяются, если различия в значениях рКb, последовательных ступеней диссоциации основания составляют не менее 4 единиц, как и в случае титрования раствс-ров полиосновных кислот раствором сильного основания.
Ошибки к-осн титр-я: 1)ошибка измерения (погрешность бюретки,пипеток)Если раствор отбирают с помощью бюретки, то проводят два измерения объема раствора в бюретке: до и после отбора раствора. Случайная ошибка каждого такого измерения при использовании обычных лабораторных бюреток составляет примерно ±(0,01—0,02) мл. Если объем отобранного раствора равен V, то максимальная случайная относительная ошибка е измерения объема, взятого для титрования, составит (в процентах):έ = ±ν*100%/V,где ν = 0,02 + 0,02 = 0,04 мл. При объеме отобранного раствора V = 20 мл величина максимальной относительной ошибки измерения объема раствора с помощью бюретки составит έ= ±0,04 • 100%/20 =0,2%.
Величину έ можно уменьшить, если увеличить объем V отбираемого раствора.
2)методические ошибки3)систематические ошибки (неправ. Подбор индикатора,несовпадение точки эквив-ти и конечной точки титрования) а)индикаторные-разница количества титранта,найденная в конечной точке титр-я и кол-ва титранта в т.экв.
а.1.)водородная ошибка (Х н3о+,Хн+)-связана с перетитровыванием р-ра сильной кислотой(тогда ошибка +) или недотитров-ем (-)Хн3о+=а/а*100%
а-кол-во избыточных эквивалентов ионов Н+ к общ кол-ву эквивалентов
с=n/V
а′=Сн3о+ *V
а=Сн3о+ * V(а+в)=сн3о+ * (Va+Vb)
-lg [H+]=pH
[H+]=C н3o+=10(в степ – рН)
Подставляем в наше вражение
Х н3о+= +-(10-pT)*(Va+Vb)/Cb*Vb)*100%
b-кислота а-щелочь.pT-показ титр-я инд
а.2.) гидроксидная ошибка(основная)-связана с избыт.кол-вом ОН групп при титр-ии сильным основанием,либо с недотитр-тью р-ром основания
а.3.)кислотная ошибка-вызвана присутствием некот.кол-ва недотитр-ой кислоты в конечной точке тир-я(слабой кислотой)
6. Окислительно – восстановительное титрование. Сущность метода. Классификация редокс – методов. Условия проведения окислительно – восстановительного титрования. Требования, предъявляемые к реакциям. Виды окислительно – восстановительного титрования (прямое, обратное, заместительное). Примеры окислительно – восстановительных индикаторов. Формулы, переход окраски в точке эквивалентности.
Окислительно – восстановительное титрование(редоксиметрия, оксидиметрия.)
К окислительно-восстановительным, относят обширную группу методов титриметрического анализа, основанных на протекании окислительно-восстановительных реакций. В окислительно-восстановительном титровании используются различные окислители и восстановители. При этом возможно определение восстановителей титрованием стандартными растворами окислителей и наоборот, определение окислителей стандартными растворами восстановителей. Благодаря большому разнообразию окислительно-восстановительных реакций этот метод позволяет определять большое количество самых разнообразных веществ, в том числе и тех которые непосредственно не проявляют окислительно-восстановительных свойств. В последнем случае используется обратное титрование. Например, при определении кальция его ионы осаждают оксалат – ионом
Ca2+ + C2O42- CaC2O4
Избыток оксалата затем оттитровывают перманганатом калия.
Окислительно-восстановительное титрование имеет ещё ряд достоинств. Окислительно-восстановительные реакции протекают достаточно быстро, что позволяет проводить титрование всего за несколько минут. Многие из них протекают в кислой, нейтральной и щелочной средах, что значительно расширяет возможности применения данного метода. Во многих случаях фиксирование точки эквивалентности возможно без применения индикаторов, поскольку применяемые растворы титрантов окрашены (KMnO4, K2Cr2O7) и в точке эквивалентности окраска титруемого раствора изменяется от одной капли титранта. Основные виды окислительно-восстановительного титрования различают по окислителю, используемому в реакции.
Окислительно-восстановительное титрование (редоксиметрию) в зависимости от природы реагента разделяют на перманганато-, дихромато-, цери, иодо-, бромато- и иодатометрию. В их основе лежит протекание окислительно-восстановительной реакции, суть которой заключается в передаче электрона от восстановителя к окислителю.
Виды ОВ титрования:
Прямое титрование заключается в том, что раствор определяемого вещества А титруют стандартным раствором титранта В. Способом прямого титрования титруют растворы кислот, оснований, карбонатов и т.д.
Обратное титрование применяют в тех случаях, когда прямое титрование не применимо: например, из-за очень низкого содержания определяемого вещества, невозможности определить точку эквивалентности, при медленном протекании реакции и т.д. В ходе обратного титрования к аликвотной части определяемого вещества А приливают точно измеренный объём стандартного раствора вещества В, взятый в избытке. Непрореагировавший избыток вещества В определяют титрованием стандартным раствором вспомогательного вещества С. По разности исходного количества вещества В и его количества, оставшегося после протекания реакции, определяют количество вещества В, вступившее в реакцию с веществом А, исходя из которого и рассчитывают содержание вещества А.
Косвенное титрование или титрование по заместителю. Основано на том, что титруют не само определяемое вещество, а продукт его реакции со вспомогательным веществом С.
А + С D
Вещество D должно образовываться строго количественно по отношению к веществу А. Определив cодержание продукта реакции D титрованием стандартным раствором вещества В, по уравнению реакции рассчитывают содержание определяемого вещества А.
Индикатор |
Окраска |
|
окисл. ф, |
восст. ф. |
|
2,2'-Дипиридил |
голубая |
красная |
Фенилантраниловая кислота |
красно-фиолетовая |
бесцветная |
Дифениламин |
сине-фиолетовая |
бесцветная |
Тионин |
фиолетовая |
бесцветная |
Индиго-тетрасульфоновая кислота |
синяя |
бесцветная |
Индиго-5,5'-дисульфонат натрия |
синяя |
желтая |
Метиленовый голубой |
синяя |
бесцветная |
Сафранин Т |
коричневая |
бесцветная |
7. Кривые окислительно – восстановительного титрования, ошибки, их происхождения, расчет, устранение. Перманганатометрия. Сущность метода, условия проведения титрования, титрант, его приготовление, стандартизация, установление точки эквивалентности. Применение перманганатометрии.
Кривые окислительно-восстановительного титрования
Кривые
редокс-титрования показывают изменение
окислительно-восстановительного
потенциала в процессе титрования: Е =
ƒ(VPB),
(рис. 2.7) В окислительно-восстановительном
титровании участвуют две редокс-системы
– титруемого вещества и титранта.
Потенциал каждой из них можно рассчитать
по уравнению Нернста, используя
соответствующую полуреакцию. После
добавления каждой порции титранта в
растворе устанавливается равновесие
и расчет потенциала можно вести по
любой из этих пар. Удобнее рассчитывать
потенциал для того вещества, которое
в данный момент титрования находится
в избытке в титруемом растворе, т.е. до
точки эквивалентности рассчитывают
потенциал по полуреакции с участием
титруемого вещества, а после точки
эквивалентности – по полуреакции с
участием титранта. До начала титрования
считают, что для титруемого вещества
концентрации окисленной и восстановленной
форм отличаются в 1000 или 10000 раз. В точке
эквивалентности в одинаковых количествах
присутствуют обе сопряженные формы
окислителя и восстановителя, поэтому
окислительно-восстановительный
потенциал можно рассчитать как сумму
потенциалов: 
Преобразуя уравнение , получим:
![]()
где n1, n2 – число электронов, участвующих в полуреакциях окисления и восстановления соответственно; Е01, Е02 – стандартный редокс-потенциал для окислителя и восстановителя соответственно.
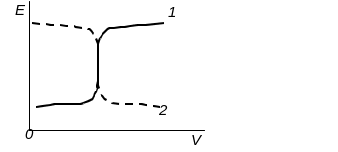
Рис. Кривые титрования в методе редоксиметрии:
1 –восстановитель титруют окислителем; 2 – окислитель титруют восстановителем
Вблизи точки эквивалентности на кривой титрования наблюдается скачок потенциала, величина которого тем больше, чем больше разница между Е0ок-ля и Е0в-ля. Индикаторное титрование возможно, если ЭДС = Е0ок-ля – Е0в-ля ≥ 0,4 В. Если ЭДС = 0,4 – 0,2 В можно использовать инструментальное титрование, где точку эквивалентности фиксируют с помощью приборов. Если ЭДС < 0,2 В прямое редоксиметрическое титрование невозможно. На величину скачка значительно влияет уменьшение концентрации одного из компонентов редокс-пары. Это порой используют для увеличения скачка на кривой титрования, что бывает необходимо при выборе индикатора.
Например, если Fe2+ титруют каким-либо окислителем, для расчета редокс-потенциала до точки эквивалентности используют редокс-пару Fe3+/Fe2+. Уменьшить начальный потенциал можно, связав ионы Fe3+ в какой-либо малодиссоциирующий комплекс, добавлением, например, фторидов или фосфорной кислоты. Так поступают при определении Fe2+ дихроматометрией. Скачок наблюдается в пределах 0,95 – 1,30 В. Чтобы проводить титрование в присутствии редокс-индикатора дифениламина (Е0 = 0,76 В), необходимо сдвинуть скачок в сторону меньших значений потенциала. При добавлении указанных комплексообразователей скачок находится в пределах 0,68 – 1,30 В. Потенциал перехода окраски дифениламина при этом находится в пределах скачка и его можно использовать для титрования Fe2+. Величина скачка зависит и от рН среды, в которой проводится реакция. Например, для полуреакции: MnO4 + 8H+ + 5e– → Mn2+ + 4H2O потенциал системы
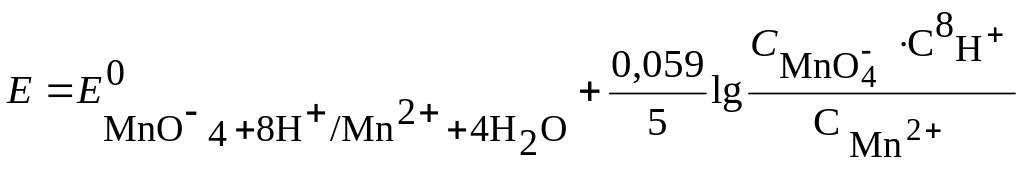 будет
увеличиваться с уменьшением рН среды,
что повлияет на величину скачка на
кривой титрования. Кривые редокс-титрования
несимметричны относительно точки
эквивалентности, если число электронов,
участвующих в полуреакциях окисления
и восстановления не равны между собой
(n1
≠
n2).
Точка эквивалентности в таких случаях
сдвинута в сторону Е0
того вещества, у которого n
больше. При титровании смесей окислителей
или восстановителей на кривой титрования
может быть несколько скачков, если
разность между редокс-потенциалами
соответствующих окислительно –
восстановительных пар достаточно
велика, в этом случае возможно раздельное
определение компонентов смеси.
будет
увеличиваться с уменьшением рН среды,
что повлияет на величину скачка на
кривой титрования. Кривые редокс-титрования
несимметричны относительно точки
эквивалентности, если число электронов,
участвующих в полуреакциях окисления
и восстановления не равны между собой
(n1
≠
n2).
Точка эквивалентности в таких случаях
сдвинута в сторону Е0
того вещества, у которого n
больше. При титровании смесей окислителей
или восстановителей на кривой титрования
может быть несколько скачков, если
разность между редокс-потенциалами
соответствующих окислительно –
восстановительных пар достаточно
велика, в этом случае возможно раздельное
определение компонентов смеси.
ПЕРМАНГАНАТОМЕТРИЯ
Перманганатометрия - метод, основанный на использовании калия перманганата в качестве титранта для определения соеди-нений, которые обладают восстановительными своиствами.
Продукты восстановления перманганат-ионов могут быть различ-ными в зависимости от рН среды:
Ø в сильнокислой среде
+ 5е + МnО4- + 8Н+ ↔ Мn2+ + 4Н2О Е0 = 1,51 В
Ø слабокислой или нейтральной среде
+ 3е + МnО4- + 4Н+ ↔ МnО2↓ + 2Н2О Е0 = 1,69 В
Ø слабощелочной среде
+ 3е + МnО4- + 2Н2О ↔ МnО2↓ + 4ОН- Е0 = 0,60 В
Для анализа чаще всего используют окислительные свойства МnО4- - ионов в сильнокислой среде, так как продуктом их восстано-вления в этом случае являются бесцветные ионы Мn 2+ (в отличие от бурого осадка МnО2), которые не мешают наблюдать изменение окраски титруемого раствора от избыточной капли титранта. Необходимое значение рН среды создают с помощью раствора серной кислоты. Другие сильные минеральные кислоты не исполь-зуют. Так, азотная кислота сама обладает окислительными свой-ствами, и в ее присутствии становится возможным протекание побочных реакций. В растворе хлороводородной кислоты (в при-сутствии следов Fe2+) происходит реакция окисления хлорид-ионов. Титрант метода - раствор 0,1 * (0,05) моль/дм3 калия перманганата - готовят как вторичный стандартный раствор и стандартизуют по стандартным веществам: щавелевой кислоте, натрия оксалату, мышьяка (ΙΙΙ) оксиду, соли Мора (NH4)2Fe(SО4)2 ∙ 6Н2О и др.
Титрованный раствор калия перманганата по точной навеске кристаллического препарата приготовить невозможно, так как в нем всегда содержатся некоторое количество МnО2 и другие продукты разложения. Перед установлением точной концентрации раствор КМnО4 выдерживают в темной склянке в течение 7-10 дней. За это время происходит окисление восстановителей, присутствие которых в дистиллированной воде полностью исключить не удается (пыль, следы органических соединений и т. п.). Для ускорения этих процессов раствор калия перманганата иногда кипятят. Необходимо учитывать, что вода обладает окислительно-восстановительными свойствами и может восстанав-ливать перманганат. Эта реакция идет медленно, но МnО2 и прямой солнечный свет катализируют процесс разложения КМnО4, поэтому через 7-10 дней осадок МnО2 необходимо удалить. Раствор КМnО4 обычно осторожно сливают с осадка или фильтруют через стеклянный фильтр. Приготовленный таким образом раствор КМnО4 не слишком низкой концентрации (0,05 моль/дм3 и выше) и не изменяет титр продолжительное время. Титр раствора калия перманганата чаще всего устанавливают по безводному натрия оксалату Na2C2O4 или щавелевой кислоте Н2С2О4 ∙ 2Н2О:
МnО4- + 5НС2О4- + 11H+ ↔ 2Мn2+ + 10СО2 + 8Н2О
Первые капли перманганата даже в горячем растворе обесцве-чиваются очень медленно. В ходе титрования концентрация ионов Мn2+ возрастает, и скорость реакции увеличивается. Титр перманга-ната калия можно установить также по мышьяка (ІІ) оксиду или металлическому железу. Использование для установления титра металлического железа особенно целесообразно, если в дальнейшем предполагается перманганатометрическое опреде-ление этого элемента.
В пермаганатометрии применяют также растворы восстано-вителей – соли Fe (ІI), щавелевую кислоту и некоторые другие - для определения окислителей методом обратного титрования. Соеди-нения Fe (ІІ) на воздухе медленно окисляются, особенно в нейт-ральном растворе. Подкисление замедляет процесс окисления, однако обычно рекомендуют перед применением раствора Fе (II) в анализе проверить его титр. Оксалаты и щавелевая кислота в растворе медленно разлагаются:
Н2С2О4 ↔ СО2↑ + СО↑+ Н2О
Этот процесс ускоряется на свету, поэтому растворы окса-латов рекомендуется хранить в темных склянках. Подкисленные растворы оксалатов более устойчивы, чем нейтральные или щелочные.
В перманганатометрии часто обходятся без применения специального индикатора, так как сам перманганат имеет интенсивную окраску, а его избыточная капля вызывает появление неисчезающей в течение 30 с розовой окраски pacтвора. При титровании разбавленными растворами применяют редокс-индикаторы, такие как дифениламинсульфокислота или ферроин (координационное соединение Fe (ІІ) с 1,10-фенантролином). Определение конечной точки титрования выполняют также потенциометрическим или амперометрическим методами.
Перманганатометрическим методом можно определить:
Ø восстановители Н2О2, NО2, C2О42-, Fe2+ и пр.,
Ø Са 2+, Ва2+ и другие катионы в различных препаратах;
Ø МnО2, РbО2, K2Cr2O7, персульфаты и другие окислители обратным титрованием. Вторым стандартным раствором в этом случае является раствор восстановителя (чаще - щавелевой кислоты или соли Мора). При этом окислители восстанавливают титрованным раствором щавелевой кислоты или соли Мора, избыток которых оттитровывают раствором калия перманганата.
Например, при анализе свинца диоксид пробу растворяют в сернокислом растворе щавелевой кислоты:
МnО2 + НС2О4- + 3H+ ↔ Мn2+ + 2 СО2↑ + 2Н2О
и избыток щавелевой кислоты оттитровывают калия перманганатом.
Перманганатометрически можно определить ионы, не обладающие окислительно-восстановительными свойствами (титрование заместителя). Этим методом могут быть определены, например, катионы кальция, стронция, бария, свинца, цинка и другие, которые образуют малорастворимые оксалаты.
Анализ органических соединений. Окисление органических соединений калия перманганатом происходит с небольшой скоро-стью, что сдерживает практическое применение этого метода для анализа органических веществ. Тем не менее некоторые органиче-ские вещества можно с успехом определять этим методом, исполь-зуя восстановление MnO4- в щелочной среде. Органические соеди-нения при этом обычно окисляются до карбоната. По окончании peaкции восстановления перманганата в щелочной среде раствор подкисляют и титруют MnO4- раствором железа (ІІ) или другого подходящего восстановителя. Так определяют, например, метанол, который в щелочной среде окисляется калия пермаганатом по схеме:
СН3ОН + 6MnO4- + 8ОН- ↔ СО32- + 6MnO42- + 6Н2О
Этим методом можно определить также муравьиную, винную, лимонную, салициловую и другие кислоты, глицерин, фенол, формальдегид и другие органические соединения.
Перманганатометрия является фармакопейным методом анализа.
8. Дихроматометрия. Сущность метода, условия проведения титрования, титрант, его приготовление, установление точки эквивалентности. Иоди – Иодометрическое титрование. Сущность метода, условия проведения титрования, титрант, его приготовление, установление точки эквивалентности.
Дихроматометрия - метод определения, основанный на окислении веществ дихромат-ионами. В его основе лежит полуреакция:
+ 6е + Сr2О72- + 14Н+ ↔ 2Сг3+ + 7Н2О Е0 = 1,33 В;
f (К2Сr2О7) = 1/6.
в кислой среде К2Сr2О7 является сильным окислителем, следова-тельно, этим методом возможно определение целого ряда неорганических и органических восстановителей, например Fe2+, [Fe(CN)6]4-, SO32-, метанола, аскорбиновой кислоты и др.
Кислую среду при титровании обычно создают с помощью раст-воров серной или ортофосфорной кислот. Можно использовать и раствор НСl при ее концентрации, не превышающей 2 моль/дм3, так как в этих условиях хлорид - ионы не окисляются дихромат- иона-ми до хлора.
Титрантом метода является раствор 0,1 моль/дм3 К2Сr2О7.
Поскольку К2Сr2О7 является стандартным веществом, из него готовят титрант первичной стандартизации.
Определение конечной точки титрования в этом методе проводят следующим образом:
Ø без индикатора - по изменению окраски титруемого раствора при добавлении избытка титранта (переход зеленого цвета Сг3+ в желто-зеленый). Метод является ненадежным;
Ø с помощью редокс-индикаторов, таких как дифениламин, фенилантраниловая кислота, дифениламиносульфокислота и др.;
Ø инструментальными методами - потенциометрией.
При определении восстановителей, например Fe2+, протекает следующая реакция:
- е + Fe2+ ↔ Fe3+ 6 Е0 = 0,771 В;
+ 6е + Сr2О72- + 14Н+ ↔ 2Сг3+ + 7Н2О 1 Е0 = 1,33 В
Fe2+ + Сr2О72- + 14Н+ → 6Fe3+ + 2Сг3+ + 7Н2О
f (Fe2+) = 1
В качестве индикатора используют натрия фениламиносульфонат, при этом окраска раствора в момент эквивалентности изменяется от зеленой до фиолетовой.
Растворы калия дихромата следует хранить в закрытой посуде в темном месте. В этих условиях титр раствора остается стабильным в течение продолжительного времени.
Иодометрия – один из окислительно-восстановительных методов объемного анализа, основан на использовании окислительно-
восстановительных свойств иода.
Атомы иода, принимая электроны от веществ – восстановителей, ведут себя в реакциях как окислители: I2o +2е → 2 I-
Анионы иода, напротив, легко отдают свои электроны веществам-окислителям и, следовательно, играют в реакциях роль восстановителей: 2I- -2e → I2o
Эти окислительно-восстановительные свойства иода и его ионов лежат в основе иодометрии.
Например, при взаимодействии иода с тиосульфатом натрия происходит реакция:
I2 + 2Na2S2O3 → 2NaI + Na2S4O6 – тетратионат натрия
S2O3 2- - 2е → S4O62- Z(Na2S2O3) = 1
I2 +2е → 2 I- Z (I2) = 2
Внешним признаком рассмотренной реакции является обесцвечивание бурого раствора иода. Точка эквивалентности при титровании устанавливается с помощью крахмала, играющего в этом случае роль индикатора (переход от синего к бесцветному).
Иодометрия чаще применяется для количественного определения окислителей. Титрование ведется, как правило, методом заместителя.
Титрование заместителя применяют, когда нет подходящей реакции или индикатора для прямого титрования.
При титровании заместителя анализируемое вещество (А) и рабочее вещество (Р) между собой непосредственно не взаимодействуют. Вначале одно из них, например, А, взаимодействует с каким-то третьим веществом (В), взятым в избытке, а получившийся при этом взаимодействии продукт (Пзам.) (он и есть заместитель) оттитровывается рабочим раствором (Р).
Т.е. количество вещества эквивалента анализируемого раствора равно количеству вещества эквивалента рабочего раствора, хотя они между собой непосредственно не взаимодействуют.
В качестве рабочего раствора в иодометрии применяют титрованный рабочий раствор тиосульфата натрия, установка точной концентрации которого осуществляются по перманганату калия тоже методом иодометрии.
Иодометрия и иодиметрия
В основе этих методов используется реакция:
![]() Восстановители,
имеющие редокс-потенциал меньше +0,534
В, способны окисляться свободным иодом.
Ионы I– способны восстанавливать
окислители, имеющие редокс-потенциал
больше + 0,534 В. Восстановители определяют
прямым или обратным (слабые восстановители)
титрованием раствором йода. Слабые
восстановители можно определять и
прямым титрованием, но для этого
предварительно необходимо понизить
потенциал их систем введением в раствор
соответствующих комплексообразователей.
Для определения окислителей используют
рабочий раствор Na2S2O3
и способ замещения.
Восстановители,
имеющие редокс-потенциал меньше +0,534
В, способны окисляться свободным иодом.
Ионы I– способны восстанавливать
окислители, имеющие редокс-потенциал
больше + 0,534 В. Восстановители определяют
прямым или обратным (слабые восстановители)
титрованием раствором йода. Слабые
восстановители можно определять и
прямым титрованием, но для этого
предварительно необходимо понизить
потенциал их систем введением в раствор
соответствующих комплексообразователей.
Для определения окислителей используют
рабочий раствор Na2S2O3
и способ замещения.
K2Cr2O7 + 7H2SO4 + 6KI → K2SO4 + Cr2(SO4)3 + 3I2 + 7H2O
I2 + 2Na2S2O3 → 2NaI + Na2S4O6
Окислитель вытесняет из KI эквивалентное количество заместителя I2, который затем оттитровывают рабочим раствором тиосульфата натрия Na2S2O3. Метод титрования раствором I2 называют иодиметрией, заместительное титрование – иодометрией. Прямая реакция восстановления иода идет быстро, но обратная реакция окисления иодид-ионов протекают медленнее.
I– – слабый восстановитель, поэтому перед титрованием анализируемую пробу с добавленным к ней КI выдерживают 5 минут в темноте. В кислой среде КI переходит в НI и на свету НI разлагается до I2, что вызовет повышенный расход раствора титранта. Обычно используют избыток KI, который необходим для лучшего растворения выделившегося I2, предупреждения его улетучивания и увеличения скорости реакции. Растворы Na2S2O3 готовят по приблизительной навеске с добавлением Na2СO3, выдерживают 7 – 10 дней, отфильтровывают выделившуюся серу и устанавливают точную концентрацию титрованием. Карбонат натрия добавляют для стабилизации тиосульфата, который легко взаимодействует с кислородом воздуха и диоксидом углерода:
2Na2S2O3 + О2 → 2Na2SO4 + 2S↓
Na2S2O3 + Н2О + СО2 → NaНSO3 + NaНСO3 + S↓
Карбонат натрия замедляет эти реакции. Рекомендуют также добавлять в раствор немного фенола или хлорамина для уничтожения тиобактерий, способствующих разложению тиосульфата натрия. В качестве установочного вещества для Na2S2O3 используют дихромат калия, раствор которого готовят из фиксанала или по точной навеске (разд 2.1, с. 23-24). Необходимо строгое соблюдение рН и температуры раствора. При повышении температуры чувствительность йод-крахмальной реакции уменьшается, кроме того, повышается летучесть иода. Реакцию ведут в слабокислой или нейтральной средах, т.к. в щелочной при рН>8 протекает побочная реакция: I2 + 2OH– → IO– + H2O + I–
Кроме окислителей и восстановителей иодометрически можно определять сильные кислоты, а также сильно гидролизующиеся соли слабых оснований. В качестве индикатора используют специфический индикатор – крахмал, который с раствором йода образует характерную синюю окраску, связанную с образованием адсорбированного комплекса (с.43). Йодометрическое определение сильных кислот H2SO4, HCl, HNO3 основано на использовании реакции, протекающей в кислой среде между йодид-ионом и иодатом калия с выделением йода.
KIO3 + 5KI + 6HCl → 6KCl + I2 + 3H2O
В нейтральной среде прекращается выделение I2. По количеству выделившегося I2 можно вычислить содержание кислоты в растворе.
При проведении анализа добавляют в избытке KIО3 (приблизительно в 3 раза) и иодид калия (в 1,5 раза) для увеличения скорости реакции и растворения образующегося йода. Выделившийся йод оттитровывают раствором тиоульфата натрия до перехода окраски из синей в бесцветную. Расчет проводят по формулам заместительного титрования (2.20 – 2.24, с.25).
Растворы I2 в иодометрии готовят по точной или приблизительной навеске, или из фиксанала. Йод легко очищается от примесей сублимацией. Следует учитывать, что йод летуч и взвешивание его надо проводить в бюксах (стеклянные стаканчики с пришлифованной пробкой). Ввиду малой растворимости йода, растворы его готовят с добавлением KI. При этом образуются растворимые в воде комплексы [I3]- или [I5]-, например: I- + I2 → [I3]-
Если необходимо, точную концетрацию раствора йода устанавливают с помощью титрованного раствора стандартного вещества оксида мышьяка As3O3 или тиосульфата натрия Na2S2O3.
В пищевой промышленности методом йодометрии определяют витамин С в овощах и фруктах, остаточный хлор в хозяйственно-питьевой воде после ее хлорирования, медь в кондитерских изделиях, сернистую кислоту в производстве сырого кукурузного крахмала, содержание хлеба в котлетном фарше, олова в консервах, общего сахара в продуктах питания.
9. Хлорйодиметрическое титрование. Сущность метода, условия проведения титрования, титрант, его приготовление, установление точки эквивалентности. Йодатометрия. Сущность метода, условия проведения титрования, титрант, его приготовление, установление точки эквивалентности. Применение методов в фарманализе.
Хлориодиметрия – метод определения воостановителей с применением титранта – р-ра монохлорида иода (ICI). Мет-д фармакопейный.
В основе лежит полуреакция: ICI- + 2е = I- + CI-. Монохлорид иода может так же воосст-ся по схеме: 2ICI + 2е = I2 + 2CI-.
Титрант – I+CI- (I+3CI-3).
Готовим р-р раствор-ие калий иодида калий иодатом:
2КIр-р + КIО3р-р + 6НСIконц→ IСI + 3КСI + 3Н2О
Отделяем I2 от всей системы с помощью хлороформа с помощью воронки. Если хлороформный слой фиолетовый, то имеется I2 , то добавляем по каплям калийиодат до обесцвечивания. Если р-р бесцветный, то берем калий иодид до фиолетового.
Стандартизируем р-р иодхлора:
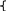 ICI
ICI
+ RI-
→ I2
+ KI
ICI
ICI
+ RI-
→ I2
+ KI
I2 + Na2S2O3 → 2NaI + Na2S4O6
Р-р не устойчив, его стандартизируем. Условия рН<7,стандартиз.соляной или серной к-ой, т.к. в щелочной среде.
Применение м-да: для опред-я фенолов, аскорб.к-ты, сульфаниламидов, парааминобензойной к-ты.
Йодатометрия.
Это определение разл.восстановителей титрованием р-ром иодата калия KIO3.
Титрант KIO3 0,1М. К раствору добавляют KI, HCl. Индикатор – крахмал, окраш-ся в фиотеовый цвет при добавлении иода.
ТЭ: KIO3 + KI + HCl а I2 ; f=1/2
Применение: опред-т в-ва облад-е восстан-ми св-ми, пр. олово, сурьма, мышьяк, иодиды, сульфиты, тиосульфаты, тиоционаты.
10. Броматометрия. Сущность метода, условия проведения титрования, титрант, его приготовление, установление точки эквивалентности. Нитритометрия. Сущность метода, условия проведения титрования, титрант, его приготовление, установление точки эквивалентности. Применение методов в фарманализе.
БРОМАТОМЕТРИЯ,
титриметрич. метод определения
восстановителей, а также органическое
соединение, вступающих с бромом в
реакции присоединения или замещения.
Основана на применении солянокислого
раствора КВrО3
с известным титром. При титровании
восстановителей бромат превращается
в бромид по схеме: BrO3-
+ 6Н+
+ 6е![]() Вr-
+ ЗН2О
(стандартный электродный потенциал +
1,45 В). Для ускорения процесса анализируемые
растворы иногда нагревают до 40-70°С или
добавляют к ним катализаторы, например
соли Hg(II), Mn(II). Конечную точку титрования
устанавливают с помощью окислит.-восстановит.
индикаторов (метилового оранжевого,
метилового красного,
Вr-
+ ЗН2О
(стандартный электродный потенциал +
1,45 В). Для ускорения процесса анализируемые
растворы иногда нагревают до 40-70°С или
добавляют к ним катализаторы, например
соли Hg(II), Mn(II). Конечную точку титрования
устанавливают с помощью окислит.-восстановит.
индикаторов (метилового оранжевого,
метилового красного,![]() нафтофлавона,
хинолинового желтого и др.), по появлению
желтой окраски брома, образующегося
при взаимодействии избытка ВrО3-
с Вr-,
а также потенциометрически или
фотометрически. Метод применяют для
определения восстановителей - As(III),
Sb(III), 71(1), Sn(II), Cu(I), Fe(II), H2O2,
гидразина, гидроксиламина, тиомочевины,
аскорбиновой и щавелевой кислот и др.
нафтофлавона,
хинолинового желтого и др.), по появлению
желтой окраски брома, образующегося
при взаимодействии избытка ВrО3-
с Вr-,
а также потенциометрически или
фотометрически. Метод применяют для
определения восстановителей - As(III),
Sb(III), 71(1), Sn(II), Cu(I), Fe(II), H2O2,
гидразина, гидроксиламина, тиомочевины,
аскорбиновой и щавелевой кислот и др.
Нитрование
органическое соединение ведут при
избытке КВr, который предварительно
добавляют в раствор титранта
(бромид-броматная смесь) или в анализируемый
раствор. При этом с определяемым
веществом взаимодействие бром,
образующийся из ВrO3-
и Вг~ по уравению: BrOJ + 5Вr-
+ 6Н+![]() ЗВr2
+ ЗН2О.
Поэтому этот вариант БРОМАТОМЕТРИЯ
(называют также бромид-броматометрией)
иногда относят к бромометрии, т.е. к
методу, основанному на применении
стандартного раствора Вг2.
ЗВr2
+ ЗН2О.
Поэтому этот вариант БРОМАТОМЕТРИЯ
(называют также бромид-броматометрией)
иногда относят к бромометрии, т.е. к
методу, основанному на применении
стандартного раствора Вг2.
Если
органическое вещество медленно
взаимодействие с выделяющимся Вr2,
к анализируемому раствору прибавляют
избыток бромид-броматной смеси и
подкисляют; после завершения бромироваия
избыток Вr2
оттитровывают раствором Na2S2O3
в присутствии KI и крахмала в качестве
индикатора или определяют обратным
арсенитометрич. методом. Последний
основан на окислении AsO2-
по схеме: AsO2-
+ ЗН2О
— 2е![]() AsO43-
+ 4H+
(стандартный электродный потенциал —
0,56 В). В этом случае к анализируемому
раствору, содержащему свободный бром,
добавляют стандартный раствор NaAsO2,
избыток которого оттитровывают броматом
в присутствии метилового оранжевого
или метилового красного. Иногда для
определения избытка брома к раствору
добавляют избыток KI и оттитровывают
выделившийся иод раствором Na2S2O3.
AsO43-
+ 4H+
(стандартный электродный потенциал —
0,56 В). В этом случае к анализируемому
раствору, содержащему свободный бром,
добавляют стандартный раствор NaAsO2,
избыток которого оттитровывают броматом
в присутствии метилового оранжевого
или метилового красного. Иногда для
определения избытка брома к раствору
добавляют избыток KI и оттитровывают
выделившийся иод раствором Na2S2O3.
Бромид-броматометрию применяют для определения фенола, крезола, анилина, резорцина, салициловой кислоты, 8-гидроксихинолина, ненасыщенных соединений (см. также Бромное число), а также для косвенного определения катионов Al, Mg, Mn(II), Ca, Ni, Co, Cu(II), Cd, Fe(III), La(III) и др., образующих с 8-гидроксихинолином нерастворимые в воде комплексные соединения, которые отделяют, растворяют в кислоте и оттитровывают выделившийся 8-гидроксихинолин.
Например, при определении стрептоцида протекает реакция (определение основано на реакции бромирования органических соединений):
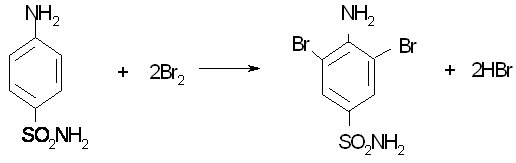
К анализируемому раствору, содержащему стрептоцид, добавляют H2SО4, раствор КВг, индикатор метиловый оранжевый и титруют стандартным раствором КВгО3 до исчезновения розовой окраски. Титрантом метода является раствор 0,1 моль/дмЗ калия бромата КВгО3. Так как КвгО3 является стандартным веществом, из него готовят титрант первичной стандартизации.
Конечную точку титрования в броматометрии определяют с помощью индикаторов метилового оранжевого или метилового красного, которые обесцвечиваются избыточной каплей титранта в результате их необратимого окисления бромом в конечной точке титрования.
НИТРИТОМЕТРИЯ.
За основу метода взяты окислительно-восстановительные, диазотирующие нитрозирующие свойства NaNО2 (в кислой среде). Поэтому им можно определить:
Ø окислители Н2О2, КМnО4, активный хлор в хлорной извести, которые окисляют нитрит-ионы до нитрат-ионов согласно уравнению:
-2е + HNО2 + Н2О ↔ NОЗ - + 3Н+ Е0 = 0,94 В;
Ø восстановители Sn2+, Fe2+, которые восстанавливают NО2 - до NO:
+ е + HNО2 + Н+ ↔ NO↑ + Н2О Е0 = 1,20 В;
Ø производные первичных и вторичных ароматических аминов.
NaNО2 (в среде НСl) может вступать в реакцию диазотирования с первичными ароматическими аминами с образованием солей диазония:
R - NH2 + NaNО2 + 2НС1 ↔ [R-N ≡N]Сl- + NaC1 + 2Н2О
Вторичные амины взаимодействуют с нитритом натрия с образо-ванием N-нитрозоаминов:
RR1NH + NaNO2 + НС1 ↔ RR1N-NO↓ + Н2О + NaC1
Титрантом метода является раствор 0,lмоль/дмЗ NaNО2, который готовят к вторичный стандартный раствор, ввиду того что кристаллический NaNО2 гигроскопичен и содержит в растворе примеси NаNО3. Приготовленный раствор NaNО2 хранят в посуде из темного стекла с притертой пробкой.
Стандартизуют раствор NaNО2:
а) по стандартным веществам (сульфаниловая кислота);
б) стандартному раствору КМnО4.
Конечную точку титрования в нитритометрии фиксируют с помощью внешних и внутренних индикаторов, а также потенциометрически.
В качестве внешнего индикатора применяют йодкрахмальную бумагу, которая в момент эквивалентности окрашивается в синий цвет.
В качестве внутренних индикаторов используют обратимые редокс-индикаторы: тропеолин 00 с метиленовым синим - окраска изменяется от фиолетовой к синей.
Метиленовый синий выполняет роль фона, на котором четче видно изменение окраски тропеолина 00. Нитритометрический метод анализа широко используют для определения многих лекарственных препаратов, содержащих первичную аминогруппу, например стрептоцида.
Нитритометрическое определение стрептоцида основано на реакции диазотирования(см. ниже большую формулу):
Поскольку взаимодействие стрептоцида с NaNО2 проходит в соотношении 1:1, то f (стрептоцида) = 1, f (NaNО2) = 1.
При выполнении реакций диазотирования необходимо соблюдать следующие условия титрования:
1. Титрование обычно выполняют на холоде (так как соли диазония разлагаются при повышенной температуре).
2. Титрование выполняют в присутствии двойного количества хлороводородной кислоты (для предотвращения протекания реакции в другом направлении).
3. Реакция диазотирования протекает медленно, поэтому для ее ускорения используют катализатор KBr.
4. При титровании желательно применять внутренние индикаторы или фиксировать конечную точку титрования потенциомет-рически.
Применение нитртометрии: для опред-ия орг-их соед-ий и неорг-их в-в, ароматических аминосоединен
11. Цериметрия. Сущность метода, условия проведения титрования, титрант, его приготовление, установление точки эквивалентности. Бромометрия. Сущность метода, титрант метода, его приготовление. Применение методов в фарманализе.
Цериметрия - метод анализа,основанный на титровании определяемого вещества раствором сульфата церия(IV)
В основе метода лежит полуреакция: С(+4)+e=C(+3)
Цериметрическое титрование проводят в кислой среде. Фактор эквивалентности церия равен единице.
Титрант-чаще сернокислый раствор сульфата церия,молярная концентрация 0,01 либо 0,1моль/л. Раствор титранта сначала готовят с приблизительной концентрацией- потом стандартизируют. 0,1 молярный готовят титрант: смешивают 100 мл воды и 28мл конц. серной кислоты,добавляют 40,4 тетрагидрата сульфата церия (CeSO4)2*4H2O.Раствор охлаждают и доводят водой до 1000мл. Стандартизацию проводят йодометрически.При этом протекает реакция Ce+4 + I- =Ce+3 +0,5 I2. затем йод оттитровывают тиосульфатом натрия.
Индикаторы метода: Кислые растворы Ce(IV) окрашены в желто-оранжевый цвет,тогда как растворы Ce(III) бесцветны.Титрование проводят в присутствии редоксиндикаторов (ферроин, орто-фенантролин, дифениламин…). Окончание титрования определяют также потенциометрически.
Можно определять: многие восстановители -ртуть, олово, мышьяк, железо, сурьму, иодиды, нитриты, тиосульфаты, пероксид водорода, щавелевую кислоту,оксалаты, аскорбиновую кислоту, фенолы, углеводы …Препараты: аминазин, вит.Е, викасол, этамзилат…
Бромометрия – метод определения восстановителей с применением в качестве реагента, взаимодействующего с определяемым веществом, раствора Br2.
Сущность метода: в основе-полуреакция: Br2 +2е= 2Br-
Титрант метода. Бромат-бромидная смесь KBrO3+KBr. Бром,образующийся при взаимодействии бромат- и бромид-ионов вступает в реакцию с определяемым веществом.
BrO(3-) + 5Br(-)+6 H(+)=3Br2+ 3H2O
При прямом титровании к кислому анализируемому раствору прибавляют в избытке бромид калия, индикатор, и титруют стандартным раствором бромата калия до исчезновения окраски индикатора в следствии взаимодействия его с бромом. При этом определяемое вещество должно бромироваться первым. После достижения ТЭ,когда всё определяемое вещество прореагирует с бромом, бромируется индикатор,вследствие чего изменяется цвет раствора.
Обратное и заместительное титрование часто проводят с иодометрическим окончанием. К сильно кислому анализируемому раствору, содержащему определяемое вещество, прибавляют в избытке точно известное количество стандартного раствора бромата калия, избыток бромида калия и оставляют смесь на некоторое время. Выделяющийся бром бромирует определяемое вещество. Затем к смеси прибавляют избыток иодида калия. Остаточный избыточный бром,не прореагировавший с определяемым веществом. вступает в реакцию с иодид-ионами: Br2 + 2I-=I2 +2 Br- Выделившийся йод титруют стандартным раствором тиосульфата натрия.
Применение: для определения различных органических веществ,в том числе препаратов-изониозида,мезатона,резорцина,салицилатов,стрептоцида,других сульфаниламидных препаратов,тимола,аминобензойных кислот,анилина,фенола и т д.
12. Комплексонометрическое титрование. Сущность метода, условия проведения титрования, понятие о комплексонатах металлов, титрант, его приготовление, установление точки эквивалентности. Влияние различных факторов на скачок на кривой титрования. Индикаторы. Меркуриметрическое титрование. Сущность метода, титрант, индикаторы. Применение методов в фарманализе.
Комплексонометрия (трилонометрия) — титриметрический метод, основанный на реакциях образования комплексных соединений ионов металлов с этилендиаминтетрауксусной кислотой и другими аминополикарбоновыми кислотами (комплексонами). Большинство ионов металлов взаимодействуют с комплексонами практически мгновенно с образованием растворимых в воде малодиссоциированных соединений постоянного состава. При комплексонометрическом титровании в результате реакции между катионом металла и комплексоном образуется комплексонат металла.
Комплексоны – это чаще всего многоосновные аминополикарбоновые кислоты и их соли, анионы которых, выступая в роли полидентатных хилатообразующих лигандов, способны образовывать со многими катионами металлов устойчивые комплексы – комплексонаты.




13. Осадительное титрование. Сущность метода, требования, предъявляемые к реакциям в методе осадительного титрования. Классификация методов по природе реагента. Кривые осадительного титрования, их расчет, построение. Индикаторы (осадительные, металлохромные, адсорбционные).
Осадительное титрование.
Метод титриметрического анализа, основанный на применении титрантов, образующих с определяемым веществом малорастворимые соединения. Конечную точку титрования обычно фиксируют с помощью индикаторов.
Например, при титровании анализируемого раствора натрия хлорида стандартным раствором нитрата серебра образуется малорастворимый осадок хлорида серебра:
Сl-+Ag+= AgСl титрование ведут до прекращения выпадения осадка. В этот момент количество титранта,пошедшего на титрование, эквивалентно количеству определяемого вещества.
Требования,предъявляемые к реакциям:
Определяемое вещество должно хорошо растворяться в воде с образованием бесцветного раствора и содержать хотя бы один ион, вступающий в реакцию осаждения с титрантом.
Реакция осаждения должна протекать строго стехиометрически. Побочные реакции или осаждение титранта либо определяемого вещества с образующимся осадком исключаются.
Реакция должна протекать практически до конца
Осадок должен выпадать быстро, при комнатной температуре, без образования перенасыщенных растворов.
Классификация методов: методы осадительного титрования обычно классифицируют по природе активного реагента, взаимодействующего с определяемым веществом:
-аргентометрия (AgNO3)
-тиоцианатометрия (NH4SCN и KSCN)
-меркурометрия (Hg2(NO3)2)
-гексацианоферратометрия (K4[Fe(CN)6])
-сульфатометрия (H2SO4)
-бариметрия BaCl2
Кривая осадительного титрования – графическое изображение изменения концентрации определяемого вещества (или титранта) в зависимости от объема прибавленного титранта ( или определяемого вещества).
Индикаторы:
Осадительные – выделяются из раствора в виде осадка в хорошо заметной форме в ТЭ или вблизи её
пример-K2CrO4 в аргентометрии по методу Мора:
Сl-+Ag+= AgСl,затем 2Ag+ + CrO4(2-)= Ag2CrO4 (красный осадок)
Т.е. титрование проводят до появления красного осадка.
Металлохромные – индикаторы, образующие с титрантом окрашенные соединения вблизи точки эквивалентности.
Пример - NH4Fe(SO4)2*12H2O железоаммонийные квасцы в аргентометрии по Фольгарду.Метод Фольгарда основан на титровании раствора, содержащего ионы серебра, стандартными растворами NH4NCS или KNCS:
Ag+ + NCS- ↔ AgNCS↓ Индикатором в этом методе являются ионы Fe3+. После осаждения ионов серебра в виде белого осадка AgNCS избыточная капля титранта реагирует с индикатором - раствором железоаммонийных квасцов NH4[Fe(SO4)2] ∙12Н2О с образованием растворимого красного комплекса:Fе3+ + 3NCS- ↔ [Fе(NСS)3]
Адсорбционные – индикаторы,адсорбция и десорбция которых осадком при осадительном титровании сопровождается изменением окраски в ТЭ или вблизи её. Это органические вещества, которые адсорбируются осадком в ТЭ и окрашивают его, а до ТЭ – не адсорбируются. Они являются слабыми протолитами кислотного или основного характера.
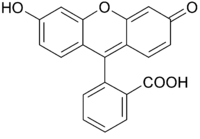 флуоресцеин
(в растворе желто-зеленый,на поверхности
осадка – розовый).
флуоресцеин
(в растворе желто-зеленый,на поверхности
осадка – розовый).
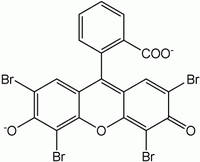 эозин
(в растворе желтовато-красный,на
поверхности осадка – красно-фиолетовый).
эозин
(в растворе желтовато-красный,на
поверхности осадка – красно-фиолетовый).




14.Аргентометрия, сущность метода, титрант, его приготовление, стандартизация. Разновидности методов (метод Гей – Люссака, Мора, Фаянса, Фишера). Индикаторы, используемые в аргентометрии. Применение аргентометрии в фарманализе.
Аргентометрия.
Сущность метода. Метод осадительного титрования, основанный на использовании стандартного раствора AgNO3 в качестве реагента-осадителя. В основе метода лежат осадительные реакции: X-+Ag+= AgX, где X-Cl,Br,I, CN, SCN
Титрование проводят в присутствии индикаторов.
Титрант-стандартный раствор AgNO3, чаще всего с концентрацией 0,05 или 0,1 моль/л.
Так как этот раствор неустойчив, его готовят вначале приблизительной концентрации, а затем стандартизуют по стандартному раствору хлорида натрия в присутствии хромата калия в качестве индикатора.
Для приготовления 0,1 моль/л раствора нитрата серебра растворяют 17г кристаллического AgNO3 в воде в мерной колбе на 1000мл и доводят обьём раствора до метки.
Стандартизация раствора AgNO3: точную навеску 0,15 кристаллического натрия хлорида, дважды перекристаллизованного из воды и выдержанного некоторое время при температуре 250-300град.цельсия, растворяют в 50 мл воды. Полученный раствор титруют стандартизуемым раствором AgNO3 в присутствие индикатора-хромата калия (K2CrO4) до появления красного осадка хромата серебра.
Расчёт концентрации и титра раствора AgNO3 проводят обычным способом, учитывая,что факторы эквивалентности AgNO3 и NaCl в данном случае равны единице:
n(AgNO3)=n(NaCl)
C(AgNO3)V( AgNO3)=C(NaCl)V(NaCl)
C(AgNO3)= C(NaCl)V(NaCl)/ V( AgNO3)
T(AgNO3)= C(AgNO3)M(AgNO3)/1000
Разновидности аргентометрии.
1.Метод Гей-Люссака- прямое титрование галогенид-ионов стандартным раствором серебра нитрата без индикатора. Окончание титрования фиксируют визуально по прекращению образования осадка сои серебра и просветлению раствора.
2. Метод Мора-прямое титрование галогенид-ионов стандартным раствором серебра нитрата в присутствии индикатора-раствора хромата калия. Применяется для определения ионов хлора и брома.
3. Метод Фаянса - прямое титрование галогенид-ионов стандартным раствором серебра нитрата в присутствии адсорбционных индикаторов – фуоресцеина, эозина и др.
4. Метод Фольгарда – обратное титрование избытка катионов серебра раствором тиоцианата аммония (NH4SCN) или тиоцианатом калия в присутствии индикатора – солей железа (III) – чаще используют железоаммонийные квасцы NH4Fe(SO4)2*12H2O. Применяется для определения галогенид-ионов, CN, SCN, S(2-),CO3(2-),C2O4(2-), AsO4(2-), CrO4(2-)…
эозин
Применение: определяют NaCl, NaBr, NaI, KI, KBr, спиртовые растворы иода, эфедрина гидрохлорид, галогенпроизводные органических веществ-бромизовала, карбромала, бромкамфоры, барбитураты.
15. Тиоцианатометрия и меркуриметрия. Сущность методов, титранты, их приготовление, стандартизация, индикаторы, применение методов в фарманализе.
Тиоцианатометрия.Сущность метода –тиоционатометримя – метод титриметрического анализа, основанный на примени стандартного раствора тиоционата аммония NH4NCS(или К NCS)в качестве реагента,взаимод.с определенным в-ом, обычно с катионами Ag+.Метод используется для определения серебра в соединениях, с примен.индикатора Фольгарда –железоаммонийных квасцов NH4Fe(SO4)2 *12H2O в азотнокислой среде.В основе метода лежит реакция: Ag+(х)+ NCS(т)→ AgSCN↓.При титровании растара вблизи ТЭ окрашивается в красный цвет вследствии образования тиоционатных комплексов железа (111).Титр. до появления желто-роз.окраски.Титрант метода.В качестве титранта метода используются водные растворы NH4NCS(или К NCS)с конц.0,1моль/л.Раствор титранта сначала готовят по приблизительной конц., а затем стандартизируют по стандартному раствору нитрата серебра.Применение.Для определения Ag, в препаратах содерж. Ag : протаргол, колларгол,в нитрате серебра.
Меркурметрия.Сущность метода. Меркуриометрия –это определение хлоридов и иодидов,титрант раствора ртуть(1)Hg2(NO3)2 в присутствии индикаторов.В основе метода-реакции образования крайне малорастворимых осадков хлорида Hg2Cl2 и иодидаHg2I2 ртути(1). Титрант метода.Титрант-водный раствор нитрата рути Hg2(NO3)2 с молярной конц.0,05моль/л. Раствор титранта сначала готовят по приблизительной конц.Полученный раствор нитрата ртути стандартизируют по станд.р-ру хлорида натрия в присут.индикатора.Индикаторы метода. Используются тиоцинатные комплексы железа(111) и дифенилкарбазон. Тиоцинатные комплексы железа(111) обрауется непосредственно титр.р-ре при введении в него тиоцианата аммония и нитрата железа (111).анализ.р-р окрашивается в красныйцвет и сохраняется до конца титрования. В процессе титрования,н-р, хлорид ионов протекает реакция: Hg2++2 Cl-→ Hg2 Cl2↓. Первая порция изб.титранта приводит к разрушению тиоцинатных комплексов железа(111) с образование растворимого тиоцината ртути(1).Красный цвет раствора исчезает. Дифенилкарбазон.(C6H5NHNH)2CO-адсорбционный индикатор, в титр.р-р прибавляется перед окончанием титрования. В близи ТЭ индикатор окрашивает осадок в синий цвет. Достоинство и недостатки по сравнению с аргентометрией. Позволяет проводить титрование сильнокислых растворов. Соединение ртути высокотоксичны, это ограничивает использование меркуриометрии.
6. Сульфатометрия, гексацианоферратометрия. Сущность методов, титранты, их приготовление, стандартизация, индикаторы, применение методов в фарманализе.
Сульфатометрия. Сущность метода. Метод определения катионов бария,путем титрования стандартным раствором серной кислоты.В основе метода -реакция осаждения катионов бария в виде малорастворимого осадка сульфата бария: Ba+2(х)+SO4-2(т)→ Ba SO4↓.Окончание титрования фиксируют индикаторным методом. Титранты метода. Титранты-стандартные растворы серной кислоты (с мол.конц.0,05моль/л), нитрата бария, хлорида бария. Раствор серной кислоты готовят как и в методе кислотно-основного титрования,растворы солей бария стандартизируют по стандартному раствору серной кислоты.Индикаторы метода. Применяют металлохромные индикаторы из группы азокрасителей –нитхромазо, ортониловыйА. Эти индикаторы окрашены в розовый цвет,а их комплексы с катионом бария в фиолетовый BaInd(фиолет) + H2SO4→BaSO4↓+ H2Ind(розовый).Достоинства. Ускоряет и упрощает проведение анализа бария или сульфатов, чем гравиметрическое определение.
Гексацианоферратометрия. Сущность метода. Метод определения катионов металлов, с использованием реагента стандартного раствора гексацианоферрат(11)калия K4[Fe(CN)6].Метод основан на реакции образования малорастворимых ферроцианидов металлов. Титрования проводят с индикаторами.Н-р,катионы Zn+2 определяют путем титрования анализир. р-ра, содер-ие катионы Zn+2, раствором ферроцианида калия в кислой среде.3 Zn+2+2K++2[ Fe(CN)6]-4→ K2Zn3[ Fe(CN)6]2↓.Титрование провоят медленно,в присутствии индикатора дифениламина(C6H5)2NH и феррицианида калия K3[Fe(CN)6].ВТЭ изменение окраски от сине-фиолетовой до салатовой.Титрант метода.Титрант –ферроцианид калия K4[Fe(CN)6].Титрант готовят сначала с приблизительно конц., азатем стандартизируют по стандартному раствору перманганата калия в сернокислой среде 5K4[Fe(CN)6]+KMnO4+4H2SO4→5K3[Fe(CN)6]+ MnSO4+ 3K2SO4+4H2O. Ind-метиленовый синий. В ТЭ изменение окраски титр.р-ра от желто-зел. До кр-коричневой.Применение. Можно измерять многие катионы:К, NH,Ag,Cu, Ca,Zn,Pb и др.
17. Титрование в неводных средах. Сущность метода кислотно – основного титрования. Классификация растворителей (протонные, апротонные). Влияние природы растворителя на силу растворенного протолита. Полнота протекания реакций в неводных растворителях. Применение кислотно – основного титрования в неводных средах.
Титрование
в неводных средах
- титрование, при котором средой служит
неводный растворитель с минимальным
содержанием воды. Неводные растворители
- обычно обезвоженные жидкости –ацетон,
диметилсульфоксид, диоксан,
к-ты(уксусная,муравьиная)и т.д. Окончание
титровании фиксируется либо индикаторным,
либо потенционаметрическими методами.
Метод фармакопейный. Силу слабых в
водных растворов кислот и оснований
можно увеличит подбором растворителя,
в котором титрование будет возможно.
Классификация
растворителей.
По способности отдавать или присоединять
протоны растворители делятся на
протонные и апротонные. Апротонные
растворители не проявляют кислотно-основных
свойств. Молекулы таких веществ не
отдают и не присоединяют протоны
(бензол,толуол,гексан). Протонные –
выраженные кислотные и основные
свойства. Способны присоединять или
отдавать протоны. Влияние
природы растворителя.
Нивелирующее действие растворителя
проявляется в выравнивании силы
растворенных в нем протолитов.
нивелирующее действие оказывают
протофильные растворители на силу
растворенных в них кислот(в водных
растворах хлорная и хлороводородные
к-ты-сильные,а уксусная к-та –слабая,
в р-ре протофильного растворителя
–жидкого аммиака – все эти 3к-ты
становятся сильными).Дифференцирующее
действие-проявляется в увел.различий
в силе растворенных в нем протолитов.
Такие расворители исп-ют для раздельного
титрования смесей к-т и оснований.Полнота
протекания реакции.
ППР (слабое основание с сильной
кислатой)Хар-ся величинй константы
равновесия К, тем больше, чем выше
константа основности Кв
титруемого
основания В и чем меньше константа
автопротолиза КSH
растворителяК= ППР(слабой
к-ты сильным основанием)хар-ся величиной
константы равновесия К, тем больше, чем
выше константа кислотной диссоциации
слабой кислоты НВ и чем меньше константа
автопротолиза КSH
растворителяК=
ППР(слабой
к-ты сильным основанием)хар-ся величиной
константы равновесия К, тем больше, чем
выше константа кислотной диссоциации
слабой кислоты НВ и чем меньше константа
автопротолиза КSH
растворителяК=![]() .
Применяют
как
основания титруются азотосодержащие
гетероцик.соединения, амиды,
амины,четвертичные аммонийные
основания,щелочные соли, н-р, в безводной
уксусной к-те можно определять многие
лекарст.в-ва –адреналин гидрат,
норадреналингидрат, амидопирин,кодеин
фсфат, метионин, резерпин и др.Как к-ты
титруются аминокислоты, карбоновые
к-ты, галогенводородные к-ты, барбитураты,
фенолы.фталазол,фурадонин.
.
Применяют
как
основания титруются азотосодержащие
гетероцик.соединения, амиды,
амины,четвертичные аммонийные
основания,щелочные соли, н-р, в безводной
уксусной к-те можно определять многие
лекарст.в-ва –адреналин гидрат,
норадреналингидрат, амидопирин,кодеин
фсфат, метионин, резерпин и др.Как к-ты
титруются аминокислоты, карбоновые
к-ты, галогенводородные к-ты, барбитураты,
фенолы.фталазол,фурадонин.
18. Инструментальные методы анализа. Общая характеристика методов анализа, их классификация, достоинства и недостатки. Оптические методы анализа. Общий принцип метода. Классификация оптических методов анализа (по изучаемым объектам, по характеру взаимодействия электромагнитного излучения с веществом, по используемой области электромагнитного спектра, по природе энергетических переходов.)
Инструментальные методы анализа. Инструментальные (физ.и физ-хим.методы)-основаны на использовании зависимости между измеряемыми физ.св-ми вещест и их качественными и количественным составом.Классификация. Основана на учете измеряемых физ. И физ-хим.свойств веществ или изучаемой системы.Оптические методы –основаны на измерении оптических свойств в-в.Хроматографические-основаны на использовании способности различных веществ к избирательной сорбции.Электрохимические методы –на измерении электрохимических свойств систем.Радиометрические-измерение радиоактивных свойств веществ.Термические методы –на измерении тепловых эффктов,соответствующих процессов.масс-спектрометрические методы-изучение ионизированных фрагментов-в.Достоинства.Низкий предел обнаружения(1-10-9мкг),малая предельная конц.(до≈10-12г/мл) определяемого вещества,высокая чуствительность,высокая селективность(избират)методов, малая продолжительность проведения анализов, возможность их автоматизации и компьютиризации. Недостатки.Иногда воспроизводимость результатов оказывается хуже, чем при использовании классических химических методов, погрешности определений часто составляют около ±5%, а в классическом методе анализа они обычно не превышают±(0,1-0,5)%, сложность применяемой аппаратуры,ее высокая стоимость.Оптические методы анализа основаны на измерения оптических свойств вещества (испускание, поглощение, рассеяние, отражения, преломлении, поляризация света),проявляющие при взаимодействии электромагнитного излучения с в-ом.Классификация. 1.По изучаемым объектам-атомный и молекулярный спектральный анализ 2. По хар-ру взаимодействия электромагнитного излучения в в-ом(атомно-адсорбционный, эмиссионный, пламенная фотометрия, молек.абсорбцюанализ,люминесцентный, спектральный, рефрактометрический и др) 3.По области используемого электромагнитного спектра(спектроскопия –в ближней УФ области, инфракрасная спектроскопия(изучающая участок электромагнитного спектр в интервале 0,76-1000мкм))4.По природе энергетических переходов(спектры-электронные,колебательные,вращательные).
19. Молекулярный спектральный анализ в ультрафиолетовой и видимой области спектра. Сущность метода, цвет и спектр. Закон Бугра – Ламберта – Бера. Оптическая плотность и светопропускание. Коэффициент поглощения и коэффициент погашения. Молярный и удельный показатель поглощения. Понятие о происхождении электронных спектров поглощения.
Молекулярный
спектральный анализ основан
на использовании способности веществ
селективно(при строго определенных
длинах волн) поглощать электромагнитную
энергию в видимой и УФ-области спектра.Цвет
и спектр.Спектр
поглощения вещества в видимой
области(≈400-760нм.) и его цвет, воспринимаемый
человеческим глазом, связаны между
собой.Цвет-свойство света вызывать
определенное зрительное ощущение в
соответствии со спектральным составом
отражаемого или испускаемого
излучения.Существуют 7 основных цвета
спектра(КОЖЗГСФ) и множества различных
оттенков между ними.Изменение цвета
в-ва в последовательности
жел→оран→крас→пурпур→синий→сине-зел
наз-ют углублением цвета, изменение
цвета в обратном порядке-повышение
цвета.При проведенииколич.анализа
оптическими методами часто имеют дело
с бесцветными средами, т.е. не поглощающими
видимый солнечный свет, в таких случаях
проводят фотометрическую реакцию, в
рез-те которой получают окрашенные
продукты реакции.
Зако́н
Бугера-Ламберта-Бера
закон,
определяющий ослабление параллельного
монохроматического
пучка света
при распространении его в поглощающей
среде.Закон выражается следующей
формулой:![]() ,где
I0
— интенсивность
входящего пучка, l
— толщина слоя вещества, через которое
проходит свет, kλ
— коэффициент поглощения (не путать с
безразмерным показателем поглощения
κ,
который
связан с kλ
формулой kλ
= 4πκ / λ, где
λ
- длина
волны).Показатель поглощения характеризует
свойства вещества и зависит от длины
волны
λ поглощаемого
света. Эта зависимость называется
спектром
поглощения вещества.
Оптическая плотность-
мера поглощения света прозрачными
объектами или отражения света
непрозрачными объектами. Вычисляется
как десятичный логарифм отношения
потока излучения падающего на объект,
к потоку излучения прошедшего через
него (отразившегося), т. е. это есть
логарифм от величины, обратнойкоэффициенту
пропускания (отражения).
,где
I0
— интенсивность
входящего пучка, l
— толщина слоя вещества, через которое
проходит свет, kλ
— коэффициент поглощения (не путать с
безразмерным показателем поглощения
κ,
который
связан с kλ
формулой kλ
= 4πκ / λ, где
λ
- длина
волны).Показатель поглощения характеризует
свойства вещества и зависит от длины
волны
λ поглощаемого
света. Эта зависимость называется
спектром
поглощения вещества.
Оптическая плотность-
мера поглощения света прозрачными
объектами или отражения света
непрозрачными объектами. Вычисляется
как десятичный логарифм отношения
потока излучения падающего на объект,
к потоку излучения прошедшего через
него (отразившегося), т. е. это есть
логарифм от величины, обратнойкоэффициенту
пропускания (отражения).![]() Прибор для измерения оптической
плотности называется денситометром.
Коэффициентом светопропускания
называется доля не поглощенного потока
света, проходящего через исследуемый
раствор. Если исследуемый раствор не
поглощает света, то α=1.
Поглощение
потока света вызывает снижение α.
Коэффициент поглощения
называют молярным, если концентрация
вещества выражена в моль/л. Он представляет
собой оптическую плотность 1 М раствора
при длине кюветы 1 см. Коэффициент
поглощения обычно используют для
сравнительной оценки чувствительности
фотометрических реакций и методик: чем
выше значение , тем меньшую концентрацию
вещества можно определить. Молярный
показатель поглощения представляет
собой оптическую плотность одномолярного
раствора вещества при толщине слоя 10
мм; удельный показатель поглощения-оптическую
плотность раствора, содержащего 1 г
вещества в 100 мл раствора при той же
толщине слоя. Понятия
о происхождении электронных спектров
поглощения при
поглощении энергии электромагнитного
излучения частицы вещества(атомы, ионы,
молекулы) увел.свою энергию,т.е.переходят
в более высоколежащее энергетическое
состояние.Для каждой частицы существует
индивидуальный набор энергетич.состояний
–энерг.уровней,н-р,электр.уровней
энергии. Самый низший энергетический
уровень –ур. С наименьшей энергией
наз-ют основным, все прочие- относятся
к возбужденным.
Прибор для измерения оптической
плотности называется денситометром.
Коэффициентом светопропускания
называется доля не поглощенного потока
света, проходящего через исследуемый
раствор. Если исследуемый раствор не
поглощает света, то α=1.
Поглощение
потока света вызывает снижение α.
Коэффициент поглощения
называют молярным, если концентрация
вещества выражена в моль/л. Он представляет
собой оптическую плотность 1 М раствора
при длине кюветы 1 см. Коэффициент
поглощения обычно используют для
сравнительной оценки чувствительности
фотометрических реакций и методик: чем
выше значение , тем меньшую концентрацию
вещества можно определить. Молярный
показатель поглощения представляет
собой оптическую плотность одномолярного
раствора вещества при толщине слоя 10
мм; удельный показатель поглощения-оптическую
плотность раствора, содержащего 1 г
вещества в 100 мл раствора при той же
толщине слоя. Понятия
о происхождении электронных спектров
поглощения при
поглощении энергии электромагнитного
излучения частицы вещества(атомы, ионы,
молекулы) увел.свою энергию,т.е.переходят
в более высоколежащее энергетическое
состояние.Для каждой частицы существует
индивидуальный набор энергетич.состояний
–энерг.уровней,н-р,электр.уровней
энергии. Самый низший энергетический
уровень –ур. С наименьшей энергией
наз-ют основным, все прочие- относятся
к возбужденным.
25. Потенциометрия. Принцип метода. Определение концентрации анализируемого раствора в прямой потенциометрии. Потенциометрическое титрование. Кривые потенциометрического титрования. Сущность метода.
Потенциометрия
электрохимический метод
исследования и анализа веществ,
основанный на зависимости равновесного
электродного потенциала Е от
термодинамической активности а Эта
зависимость описывается Нернста
уравнением![]()
при потенциометр. Измерениях в электрохим.ячейке используют два электрода индикаторн.электрод ,потенциал которого зависит от конц. Опред. В-ва в анализируемом р-ре, и электрод сравнения, потенциал которого постоянен
гдe Е0 - стандар-тная ЭДС цепи; п - заряд анализируемого иона с соответствующим знаком; S - крутизна электро-дной функции индикаторного электрода, селек-тивного к однозарядному иону; аан - активность анализируемого иона.Для потенциометрических измерений применяют электрохимические цепи, содержащие два электрода: индикаторный и элек-трод сравнения. Если оба электрода погружены в анализируемый раствор, то такая цепь называется цепью без переноса. Если электрод сравнения сое-диняют с анализируемым раствором через жидко-стный контакт (солевой мостик), то цепь называ-ется цепью с переносом.В потенциометрическом анализе используют преи-мущественно цепи с пе-реносом. Схематически такую цепь изображают следующим образом: Индикаторный электрод – Анализируемый раствор- Солевой мостик- Элек-трод сравнения Индикаторным называют элек-трод, потенциал которого определяет активность анализируемого иона в соответствии с уравнением Нернста. Электродом сравнения называют элект-род, по-тенциал которого постоянен и не зависит от кон-центрации ионов в растворе. Солевой мостик служит для предотвращения смешивания анализи-руемого раствора и раствора электрода срав-нения. В качестве солевого мостика используют насыщен-ные растворы солей KCl, КNО3
26. Полярографический анализ. Общие понятия, принцип метода, полярографические кривые, потенциал полуволны, связь величины диффузионного тока с концентрацией. Количественный полярографический анализ, определение концентрации анализируемого раствора.
Полярографический анализ основан на использовании след. зависимостей между электр. параметрами,электрохим. ячейки, которая прилагается внешний потенциал, и свойствами содержащегося в ней анал. раствора : в качественном поляграфическом анализе используют связь между величенной приложенного на микроэлектроде внешнего элек.потенциала, при кот наблюдается восстановление анализируемого в-ва.В количеств. анализе используют связь между величеной диффузного электр. тока, устанавливающегося в полярографической ячейке после достяжения опред. значения элект. потенциала, и концент-ей определяемого в-ва в анализируемом р-ре.Вместо потенциала выделения на практике опред. потенциал полуволны соответствующей половине величины диффуз. тока.Полученную полярографическую кривую наз. полярограммой или волной.Количественный полярографический анализ основан на измерении диффузного тока как функции концентрации определяемого полярограф. активного в-ва деполяризатора в полярографируемом р-ре.При анализе получаемых полярограмм концентрацию определяемого вещества находят методами градуировочного графика , добавок стандарта ,стандартного р-ра.
27. Амперометрическое титрование. Сущность метода, условия проведения амперометрического титрования. Кривые амперометрического титрования. Применение метода. Кулонометрия. Принципы. Прямая кулонометрия. Сущность метода. Кулонометрическое титрование.
Амперометрическое титрование-разновидность вольфрам –перометрического метода.различают амперомет. Титрование с одним поляризуемым электродом,назыв. титрованием по предельному току, и титрование с двумя одинаковыми поляризуемыми электродами, или биамперометрическое титрование Первый метод основан на изменении тока в поляграфической ячейке в зависимости от количества прибавленного титранта при постоянном внешнем потенциале на микроэлектроде.титрование ведут на установке , состоящей из источника пост. Тока с регулируемым напряжением, к которому присоединены гальванометр и полярографическая ячейка для титрования.В полярографическую ячейку вносят анализируемый р-р, и прибавляют небольшими порциями титрант,измеряя ток .Величина тока зависит от концентрации полярографичечески активного вещества.В точке эквивалентности величена резко изменяется.По результатам амперометрического титрования строят кривые титрования. Кривая амперометрического титрования –это графическое представление изменения величены тока в зависимости от объёма прибавленного титранта.Примером может служить титрование сульфат-ионов раствором,содержащим катионы свинца.Биамперометрическое титрование- фармак. Метод;применяется в иодометрии,нитритометрии, акваметрии, при титровании в неводных средах.
Кулонометрия.основан на использовании зависимости между массой вещества, прореагировавшего при электролизе в электрохимич.ячейке , и кол-ом электричества прошедшего через ячейку этого в-ва.Главная задача кулонометрических измерений-как можно более точно определить кол-во электричества.Кулономет.анализ проводят либо в амперостатич.режиме ,т.е. при постоянном элект. Токе либо при контролируемом пост.потенциале рабочего электрода ,когда элек.ток изменяется в процессе электролиза.Прямая кулонометрия электролизу подвергают непосредст. Определяемое в-во.измеряют к-во электричества,затраченное на электролиз этого в-ва, и по ур-ю рассчитывают массу определ. в-ва.При кулонометрическом титровании определяемое в-во,находящееся в р-ре в электрохим.ячейке ,реагирует с титрантом образующемся на генераторном электроде при электролизе вспом в-ва.Окончание титрования-момент ,когда всё опред.в-во полностью прореагирует с генерируемым титрантом ,фиксирует вводя в р-р индикатор.Прикулонометрии титрант не прибавляется из бюреткив р-р.Роль титранта играет в-во непрерывно генерируемое при электрод. Р-циина генераторном электроде.
28. Гравиметрия. Основные понятия гравиметрического анализа. Классификация методов гравиметрического анализа (метод осаждения, метод отгонки, метод выделения, термогравиметрический метод). Метод осаждения. Основные этапы гравиметрического определения (краткая классификация каждого этапа).
Гравимметрия Гравиметрический метод. основан на точном измерении массы вещества известного состава, химически связанного с определяемым компонентом и выделенного в виде соединения или в виде простого вещества Метод характеризуется высокой точностью, но длителен и трудоемок. В фармацевтическом анализе его применяют для определения влажности и зольности лек препаратов. 1Расчет массы навески анализируемого вещества и ее взвешивание 2 Растворение навески 3 Создание условий осаждения 4 Осаждение (получение осажденной формы) 5Отделение осадка фильтрованием 6 Промывание осадка 7 Получение гравиметрической формы (высушивание, прокаливание до постоянной массы) 8 Взвешивание гравиметрической формы 9 Расчет результатов анализа Осажденной формой называют соединение, в виде которого определяемый компонент осаждается из раствора. Гравиметрической (весовой) формой называют соединение, которое взвешивают. Осажденная форма должна быть:достаточно малорастворимой, чтобы обеспечить практически полное, полученный осадок должен быть чистым и легко фильтрую-щимся (что определяет преимущества кристалли-ческих осадков);осажденная форма должна легко переходить в гравиметрическую форму. Основными требованиями к гравиметрической форме являются точное соответствие ее состава определенной химической формуле; химическая устойчивость в достаточно широком интервале температур, отсутствие гигроскопичности
29. Осаждаемая и гравиметрическая форма. Условия образования кристаллического и аморфного осадка, требования, предъявляемые к осаждаемой и гравиметрической форме, осадителю, промывной жидкости. Примеры гравиметрических определений.
Осаждаемая и гравиметрическая форма осн.цель при получении ос. формы –свести к минимуму потери за счет растворения осадка в моточном растворе:чтобы осадок не содержал примесей,чтобы частицы осадка были достаточно крупными Требования к осадку: опред. компанент должен переходить в осадок,не должен содержать примеси, д.б.устойчивым к внеш. Воздействиям, при высушивании или прокаливании превращ.в гравиметрич. форму, структура осадка должна обеспечивать оптим. проведение фильтрования фильтрования осадка от примесей.Условия образования осадка .Осадок образуется толькотогда когда конц.р-ра становиться выше конц.насыщ. р-ра.обычностараются проводить осаждение в таких условиях когда степень пересыщения мала.Условия получения крист. Осадков:осаждение следует вести из разбавленного анализ.р-ра разбав. р-ром осадителя.,р-росадителя прибавл. медленно,по каплям,выпавший осадокоставляют на некот. Время для созревания осадка.В гравиметрическом анализе при работе с аморф. осадками для предотвращения коллоидн р-ров, вводят электролит-коагулятори повыш. температуру .Условия получения аморфных осадков:к горячему конц. анализ. р-ру +гор. конц.р-р осадителя, при необходимости вводят электролит-коагулятор. Определяют большинство катионов металлов,анионов лек. Раст. Сырья опред. золы.
1. Аналитическая химия как наука. Основные понятия (метод, методика, качественный, количественный, структурный, элементный, функциональный). История развития аналитической химии. Применение методов в фармации, фармацевтический анализ. Аналитические признаки веществ и аналитические реакции.
АНАЛ,ХИМИЯ КАК НАУКА- это раздел химической науки, разрабатывающий на основе фундаментальных законов химии и физики принципиальные методы и приемы качественного и количественного анализа атомного, молекулярного и фазового состава вещества. Метод анализа вещества — это краткое определение принципов, положенных в основу анализа, вещества. Методика анализа — подробное описание всех условий и операций, которые обеспечивают регламентированные характеристики, в том числе__—-_ правильности и воспроизводимости, результатов анализа. Качественный химический анализ — это определение (открытие) химических элементов, ионов, атомов, атомных групп, молекул в анализируемом веществе.Количественный химический анализ — это определение количественного состава вещества, т. е. установление количества химических элементов, ионов, атомов, атомных групп, молекул в анализируемом веществе. количественный анализ вещества — это экспериментальное определение (измерение) концентрации (количества) химических элементов (соединений) или их форм в анализируемом веществе, выраженное в виде границ доверительного интервала или числа с указанием стандартного отклонения. Элементный анализ — это качественный и (чаще всего) количественный химический анализ, в результате которого определяют, какие химические элементы и в каких количественных соотношениях входят в состав анализируемого вещества.Функциональный анализ — открытие и определение различных функциональных групп, например, аминогруппы МН2, нитрогруппы N02, карбонильной С=0, карбоксильной СООН, гидроксильной ОН, ширильной СN групп и др.Молекулярный анализ — открытие молекул и определение молекулярного состава анализируемого вещества, т. е. выяснение того, из каких молекул и в каких количественных соотношениях состоит данный анализируемый объект.
Фазовый анализ — открытие и определение различных фаз (твердых, жидких, газообразных), входящих в данную анализируемую систему. ИСТОРИЯПрактическая анал хим возникла еще в глубокой древности. Как наука ан хим начала развиваться с середины XVII в. Большой вклад в развитие внес Р. Бойль, который систематизировал известные до него качественные реакции и предложил новые. Все реакции проводились в растворе.Микрокристаллоскопический метод анализа возник в России в XVIII в. благодаря трудам М В. Ломоносова и Т. Е. Ловица. Непосредственным преемником и продолжателем работ М. В. Ломоносова был русский ученый В. М. Севергин. Он обогатил аналитическую химию рядом новых реакций и приемов анализа.
Количественный анализ начал развиваться в XIX в. Большие заслуги в этой области принадлежат И. Я- Бер-целиусу и Ю. Либиху. В 20-х годах XIX в. Ж. Гей-Люссак ввел объемный метод количественного анализа. Р. Бунзен и Г. Кирхгоф в середине XIX в. разработали спектральный анализ. методы анализа:титриметрия, объёмный анализ; весовой анализ — гравиметрия, электрогравиметрия ;кулонометрия ;колориметрия; фотометрия ;
электрохимические методы и т.д. На основе методов аналитической химии (аналитики) осуществляется фармацевтический анализ — определение качества лекарств и лекарственных средств, изготовляемых промышленностью и аптеками. Фармацевтический анализ включает: анализ лекарственных препаратов, лекарственного сырья, контроль производства лекарств, токсикологический анализ (определение содержания токсических веществ) в объектах растительного и животного происхождения, судебно-химический анализ. Фармацевтический анализ обычно проводится в контрольно-аналитических лабораториях институтов, химико-фармацевтических, заводов, фабрик и т. д. Для контроля качества лекарственных средств используют фармакопейные методы анализа. Аналитические признаки — такие свойства анализируемого вещества или продуктов его превращения, которые позволяют судить о наличии в нем тех или иных компонентов. Характерные аналитические признаки — цвет, запах, угол вращения плоскости поляризации света, радиоактивность, способность к взаимодействию с электромагнитным излучением (например, наличие характеристических полос в ИК-спектрах поглощения или максимумов в спектрах поглощения в видимой и УФ-области спектра) и др.
Аналитическая реакция — химическое превращение анализируемого вещества при действии аналитического реагента с образованием продуктов с заметными аналитическими признаками. В качестве аналитических реакций чаще всего используют реакции образования окрашенных соединений, выделение или растворение осадков, газов, образование кристаллов характерной формы, окрашивание пламени газовой горелки, образование соединений, люминесцирующих в растворах.
2. Подготовка образца к анализу. Средняя проба, отбор средней пробы. Чувствительность химических реакций. Характеристика чувствительности аналитических реакций (обнаруживаемый минимум, предельное разбавление, предельная концентрация, минимальный объем предельно разбавленного раствора. Способы повышения чувствительности.
Если количественные измерения проводят в растворе, образец растворяют в подходящем растворителе; при этом концентрацию образца подбирают так, чтобы она находилась в пределах применимости метода. Иногда приходится выделять определяемое вещество из смеси, поскольку многие методы анализа неспецифичны и даже неселективны. Специфичным называют метод, при помощи которого определяется только конкретное вещество, а селективным - предпочтительный для данного вещества метод, пользуясь которым можно определять и другие вещества. Специфичных методов очень мало, селективных - значительно больше. Например, высоко-селективны масс-спектрометрия и иммунологический анализ. Средняя проба — это небольшая представительная часть вещества, состав и свойства которой идентичны составу и свойствам всей массы анализируемого вещества.Способ отбора средней пробы зависит от природы анализируемого вещества, его агрегатного состояния, однородности. Отбор средней пробы. Состав отобранной средней пробы должен приближаться к среднему химическому составу большого количества исследуемого материала. Особенно трудно отобрать среднюю пробу тогда, когда в нашем распоряжении будет находиться твердое вещество с явно выраженной неоднородностью. Если средняя проба будет отобрана неправильно, самый точный анализ не будет отражать химического состава исследуемого материала. Поэтому государственные стандарты и технические условия предусматривают правила отбора средних проб. Но отобранная одним из указанных способов первичная средняя проба, как правило, очень велика и неоднородна. Поэтому ее измельчают, перемешивают и сокращают. Сокращение проводят способом квартования. Если же необходимо найти содержание химического элемента или какой-либо составной части химического соединения, необходимо предварительно удалить примеси. Для очистки от примесей разработаны различные методы. Как для очистки, так и для разделения веществ пользуются перекристаллизацией, Чувствительность анал. реакций определяет возможность обнаружения вещества (ионов, молекул) в растворе. Предельное разбавление Vlim — максимальный объем раствора, в котором может быть однозначно обнаружен 1 грамм данного вещества при помощи данной аналитической реакции( выражается в мл/г). Предельная концентрация clim (или cmin) — наименьшая концентрация, при которой определяемое вещество может быть обнаружено в растворе данной аналитической реакцией,выражается в г/мл. Предельная концентрация и предельное разбавление связаны соотношением clim=1/Vlim. Минимальный объем предельно разбавленного раствора Vmin — наименьший объем анализируемого раствора, необходимый для обнаружения открываемого вещества данной аналитической реакцией. Выражается в мл. Предел обнаружения (открываемый минимум) т (в мкг) — наименьшая масса определяемого вещества, однозначно открываемого данной аналитической реакцией в минимальном объеме предельно разбавленного раствора. Повысить чувствительность реакции можно также, применяя химически чистые реактивы, выпаривание растворов ( для увеличения концентрации), предварительное осаждение и последующее растворение полученного малорастворимого соединения в подходящем растворителе, экстрагирование соединений органическими растворителями и другие специальные методы. Чувствительность аналитических реакций определяется также временем, в течение которого протекает реакция. Реакции, в которых реактив реагирует с обнаруживаемым ионом за более короткий промежуток времени, считаются более чувствительными.
3. Избирательность химических реакций. Классификация реагентов в аналитических реакциях (привести примеры специфичных, избирательных и групповых реагентов). Способы увеличения избирательности. Привести примеры.
Для характеристики этого свойства обычно перечисляют химический элементы (или химический соединение), которые взаимодействие с данным ОР при определенных условиях. Чем меньше таких элементов (соединение), тем выше избирательность. Избирательность можно повысить, изменяя условия выполнения реакции или модифицируя структуру самого реагента, т.е. синтезируя аналоги данного ОР. Первый путь сводится к подбору оптим. условий выполнения реакции, в частности рН среды. Универс. способ повышения избирательности анализа - применение различные маскирующих веществ, которые связывают компоненты, сопутствующие определяемому. Иногда при определении ионов металлов в реакционное систему вводят дополнительно химический соединение, что приводит к образованию смешанно-лигандного комплексного соединения (т. е. соединение, содержащего различные лиганды), характерного только для определяемого элемента. Специфические реагенты и реакции позволяют обнаруживать данное вещество или данный ион в присутствии других веществ или ионов. Например на йод-крахмал,.Смесь а-нафтиламина с сульфаниловой- на нитриты, на никель-ректив Чугаева. Селективные реагенты и реакции позволяют обнаруживать несколько веществ или ионов. Так, при действии NH.SCN на смесь катионов только два катиона образуют растворимые окрашенные комплексные соединения: [Fe(SCN)6]3_ и (Co(SCN),]2. Групповые реагенты позволяют обнаруживать ионы определенной аналитической группы.(HCl-реагент на катионы IIгр-Pb+2,Hg2+2,Ag+1).