

Механізми виникнення та розвитку захворювання (Патогенез)
Ниркові АГ включають
ренопаренхіматозні;
реноваскулярні;
посттрансплантаційні.
Ренопаренхиматозні гіпертензії є найбільш частою причиною вторинних гіпертензій. Вони характеризуються гіперволемією, гіпернатріємією, збільшенням периферичного судинного опору і нормальним або пониженим серцевим викидом. Гіперволемія і гіпернатріємія є результатом прогресуючого ураження нефронів і / або інтерстиціальної тканини нирок, зниження здатності до екскреції натрію і води.
Підвищена периферична вазоконстрикція в основному пояснюється активацією ренін-ангіотензинової системи з гіперангіотензинемією, у ряді випадків – зниженням утворення і функції ниркових і судинних депресорних систем.
В цілях прискорення діагностики ренопаренхиматозних гіпертензій виділяють симетричні і асиметричні хвороби нирок.
Симетричні: первинний гломерулонефрит (гострий, підгострий, хронічний); вторинні гломерулонефрити при системних захворюваннях сполучної тканини і системних васкулітах (системний червоний вовчак, склеродермія, гранульоматоз Вегенера, вузликовий періартеріїт, синдром Гудпасчера та ін.); діабетична нефропатія, інтерстиціальний анальгетичний нефрит, полікістоз нирок.
Асиметричні: природжені аномалії нирок, рефлюксна нефропатія, пієлонефрит, солітарні кісти нирок, травма нирки, сегментарна гіпоплазія, постобструктивна нирка, однобічний туберкульоз нирки (АГ спостерігається рідко), однобічний радіаційний нефрит, пухлина нирки, ренін-утворююча пухлина.
При асиметричних нефропатіях існує можливість лікування АГ шляхом нефректомії. Нефректомія показана при важкій АГ, значному порушенні функції ураженої нирки і нормальній функції іншої нирки.
Реноваскулярна гіпертензія (РВГ) є найбільш курабельною формою вторинної гіпертензії, її частота в загальній популяції складає 0,5 %.
Причини і частота РВГ: атеросклероз - 60-70 %, фібромускулярна дисплазія 30 - 40 %, рідкісні причини (< 1 %): ниркова артеріовенозна фістула, аневризма ниркової артерії, тромбоз ниркової артерії, тромбоз ниркових вен. Інші хвороби, які можуть бути причиною РВГ (< 1 %): коарктація аорти, аневризма аорти, вузликовий періартеріїт, хвороба Такаясу, феохромоцитома, рак або метастази пухлини, нейрофіброматоз (хвороба Реклінгаузена), ниркові кісти.
У патогенезі РВГ основне значення надається хронічній стимуляції ренін-ангіотензинової системи у зв'язку з гіпоперфузією нирки на боці стенозу. Поглиблене інструментальне і діагностичне обстеження для підтвердження або виключення РВГ проводиться не всім хворим, а тільки за наявністю клінічних свідчень.
Ендокринні артеріальні гіпертензії
Зустрічаються у 0,05 % всіх хворих артеріальною гіпертензією, вони складають приблизно 15 % вторинних форм гіпертензії.
Феохромоцитома – катехоламін-утворююча пухлина нейроектодермальної тканини, яка локалізується в 85 % випадків в мозковій речовині надниркових залоз і в 15 % ‑ поза наднирковими залозами: у абдомінальному і грудному симпатичних стовбурах, органі Цуккеркандля, можливе ектопічне розташування скупчень хромафінних клітин.
Пухлина зазвичай доброякісна, але в 5-10 % випадків спостерігається злоякісна феохромобластома. У 10 % визначається генетична схильність до пухлини, при цьому характерне її поєднання з хворобами інших органів і тканин (гіперплазія щитоподібної залози, аденома паращитоподібних залоз, нейрофіброматоз, хвороба Реклінгаузена, синдром Марфана, гангліонейроматоз шлунково-кишкового тракту, ангіоматоз сітківки і головного мозку, медуллярна карцинома щитоподібної залози).
Первинний альдостеронизм. Первинний альдостеронизм характеризується автономною гіперпродукцією альдостерону. Вторинний альдостеронизм може бути при наступних ситуаціях, пов'язаних із збільшенням активності реніну плазми: есенціальна гіпертензія, ренін-секретируюча пухлина; ренопаренхімні, реноваскулярні гіпертензії; феохромоцитома; прийом діуретиків, контрацептивів; застійна серцева недостатність; гострий інфаркт міокарду.
Первинний альдостеронизм може бути класифікований по патогенетично різних формах:
первинна гіперплазія кори надниркових залоз;
альдостерон-утворююча аденома надниркової залози;
карцинома надниркової залози;
ідіопатична білатеральна гіперплазія кори надниркових залоз;
дексаметазон-пригнічуємий гіперальдостеронізм;
позанадниркова альдостерон-утворююча пухлина.
Дексаметазон-пригнічуємий гіперальдостеронизм передбачається за наявності гіперплазії надниркових залоз. Основними клінічними і біохімічними ознаками є: нормалізація АТ, рівнів альдостерону і калію після прийому дексаметазону, наявність гібридних стероїдів 18-гідроксікортизола і 18-оксікортизола.
Синдром Іценко-Кушинга (гіперкортіцизм) – це клінічний симптомокомплекс, обумовлений ендогенною гіперпродукцією або тривалим екзогенним введенням кортикостероїдів. Патогенез гіпертензії при гіперкортіцизмі багатофакторний:
мінералкортикоїдний ефект високого рівня кортизолу із затримкою натрію та рідини (зі збільшенням екскреції калію);
глюкокортикоїдна стимуляція продукції ангіотензиногену з подальшим посиленням продукції ангіотензину II;
збільшення продукції та вивільнення мінералкортикоїд-активних попередників стероїдгенезу;
збільшена активність симпатичної системи;
збільшена реактивність судин на вазопресори;
зниження депресорних чинників (кінінів, простагландинів)
Причини синдрому Іценко-Кушинга можуть бути АКТГ-залежні та АКТГ-незалежні: 1) АКТГ-залежні – хвороба Кушинга (патологія гіпофіза), ектопічний АКТГ або кортикотропін-рилізінг сіндром – 80 %; 2) АКТГ-незалежні: аденома надниркових залоз, карцинома, мікро- або макронодулярна гіперплазія – 20 %; 3) псевдокушингоїдний синдром (великі депресивні розлади, алкоголізм) – 1 %; 4) ятрогенні – гіперкортицизм унаслідок тривалого прийому глюкокортикоїдів.
Акромегалія – артеріальна гіпертензія зв'язана із затримкою натрію внаслідок гіперпродукції соматотропного гормону.
Гіпотиреоз – гіпертензія асоціюється із збільшеним рівнем діастолічного тиску внаслідок збільшеного периферичного опору.
Гіпертиреоз – артеріальна гіпертензія характеризується збільшенням систолічного тиску і зниженням діалісточного тиску. Збільшення діастолічного тиску при гіпертиреозі припускає іншу причину гіпертензії.
Первинний гіперпаратиреоїдизм в 10 разів частіше зустрічається при АГ, чим в загальній популяції, з іншого боку, АГ спостерігається у 10-70 % хворих з первинним гіперпаратиреоїдизмом (ГПТ). Можливо, що АГ у частини хворих з ГПТ пояснюється існуванням паратиреоїдного гіпертензивного чинника (ПГX), який також продукується в паращитоподібних залозах, проте поки що немає чітких доказів ендокринно-пов'язаної гіпертензії при ГПТ.
Кардіоваскулярні гіпертензії
Представлені випадками підвищення артеріального тиску, причиною яких є первинні захворювання серця і судин.
Кардіоваскулярні гіпертензії включають:
1) первинні ураження серця (гіпертензія внаслідок збільшеного серцевого викиду);
2) гіперкінетична циркуляція;
3) недостатність півмісяцевих клапанів аорти;
4) повна атріо-вентрикулярна блокада;
5) первинні ураження аорти:
атеросклероз аорти;
коарктація аорти;
стеноз дуги або перешийка аорти (рідше – грудного або черевного відділів).
З перерахованих форм гіпертензії потенційно курабельною формою є коарктація аорти.
Неврогенні гіпертензії
Термін “неврогенні гіпертензії” відображає хронічні форми гіпертензії, які обумовлені неврологічними захворюваннями.
Синдром апное під час сну. Терміном “сонне або нічне апное” визначають синдром повного припинення дихання уві сні тривалістю 10 секунд і більш. Якщо такі епізоди повторюються 5-6 разів і більш впродовж 1 години сну, говорять про синдром “сонного апное”.
Розрізняють апное центральне, обструктивне і змішане; центральне ‑ зниження центральної активації дихальних м'язових груп, обструктивне, - інспіраторна обструкція верхніх дихальних шляхів унаслідок релаксації або інактивації м'язових груп, які відповідають за розкриття екстраторакального респіраторного тракту. Найбільш частою є змішана форма, яка зустрічається у 4 % чоловіків і 2 % жінок.
При нормальному сні систолічний артеріальний тиск у людини на 5 – 14 % нижче, ніж у людини, що не спить. При синдромі апное артеріальний тиск підвищується, у деяких хворих може досягати 200-300 мм рт. ст., при частих епізодах апное може відмічатися постійна нічна гіпертензія. Указують на високу частоту стійкої гіпертензії (і в денний час) у хворих на апное під час сну, що становить 30-50 %.
При дослідженні патогенезу артеріальної гіпертензії у хворих з синдромом “сонного” апное встановлена підвищена адренергічна активність, посилення утворення ейкозаноїдів, ендотеліну, аденозіна, передсердного натрійуретичного чинника.
Епідеміологічним маркером апное під час сну є хропіння уві сні. Більшість хворих на апное під час сну мають надмірну масу тіла, частіше визначається ожиріння верхньої половини тулуба.
Встановлена схильність хворих на апное під час сну до серцево-судинної смертності, для пояснення якої висунуті наступні припущення: наявність стійкої гіпертензії, гіпертрофія лівого шлуночку, гіперсимпатикотонія; гіпоксемія під час епізодів апное може провокувати порушення ритму і провідності, гостру коронарну недостатність, інфаркт міокарду, інсульт.
Артеріальна гіпертензія, обумовлена прийомом ліків
28.24. Які існують експериментальні моделі артеріальної гіпертензії?
Жодна хвороба людини не має такої великої кількості різних експериментальних моделей, як артеріальна гіпертензія. Нині артеріальну гіпертензію вивчають на мишах, щурах, кролях, кішках, собаках, свинях, мавпах.
За методами відтворення всі моделі артеріальної гіпертензії можна поділити на кілька великих груп.
I. Порушення функції центральної нервової системи:
а) зіткнення процесів умовного збудження і гальмування, що призводить до розвитку у тварин (собак, мавп) неврозу;
б) моделювання психоемоційної напруги шляхом створення зоосоціального конфлікту (у мавп), змін біоритмів, іммобілізації тварин;
в) електрична і хімічна стимуляція лімбічних структур головного мозку.
II. Порушення мозкового крово- і лімфообігу:
а) одно- і двостороннє перев'язування сонних і вертебральних артерій, що живлять мозок (центрально-ішемічна артеріальна гіпертензія)',
б) блокада лімфовідведення по периневральних і периваскулярних лімфатичних шляхах за допомогою каоліну, що його вводять у велику цистерну мозку.
III. Порушення функції депресорних регуляторних систем:
а) двостороннє перетинання у кролів і собак депресорних і синусних нервів, у результаті чого знімаються гальмівні впливи з барорецепторів рефлексогенних зон дуги аорти і каротидного синуса (рефлексогенна гіпертензія, або гіпертензія розгальмування);
б) центральна деаферентація барорецепторів, що її викликають ушкодженням ядра солітарного тракту;
в) пригнічення синтезу простагландинів за допомогою індометацину.
IV. Порушення функції нирок:
а) звужування обох ниркових артерій або звужування однієї ниркової артерії з видаленням другої контрлатеральної нирки (реноваскулярна гіпертензія). Виникнення артеріальної гіпертензії в цьому випадку пов'язане з активацією ренін-ангіотензинної системи;
б) видалення обох нирок і переведення тварин на гемодіаліз для запобігання уремії (ренопривна гіпертензія). її розвиток пояснюють припиненням депресорних функцій нирок;
в) обгортання нирок целофаном, шовком. При цьому виникає перинефрит:' здавлюється ниркова паренхіма, розвивається венозний застій і гіпоксія нирок, активується ренін-ангіотензинна система.
V. Порушення гормонального стану:
а) введення тваринам адреналіну;
в) введення вазопресину;
в) субтотальне видалення кори надниркових залоз. При цьому відбувається посилення регенерації залозистої тканини з посиленою продукцією кортикостероїдів, особливо альдостерону (надшрнико-регенераційна гіпертензія).
VI. Порушення водно-сольового обміну:
а) введення тваринам великої кількості кухонної солі (сольова гіпертензія)',
б) введення мінералокортикоїдів (дезоксикортикостерону, альдостерону) - мі-нералокортикоїдна гіпертензія;
в) поєднане введення кухонної солі й мінералокортикоїдів.
VII. Моделі генетично обумовленої артеріальної гіпертензії. У багатьох лабораторіях світу виведено чисті лінії щурів, характерною рисою яких є гіпертензія -ознака, що спадкується. Це, зокрема, щури зі спонтанною гіпертензією (лінія Окамото - Аокі); щури, схильні до інсультів; новозеландські щури, міланські щури; щури, чутливі до сольової дієти, та ін.
Первинна артеріальна гіпертензія як мультифакторіальне захворювання; сучасні уявлення про етіологію та патогенез гіпертонічної хвороби. Роль нирок в патогенезі первинної артеріальної гіпертензії.
Артеріальна гіпертензія (АГ) - патологічний стан, який характеризується підвищенням артеріального тиску.
Усі випадки гіпертонічної хвороби підрозділяються на гіпертонічну хворобу (ГХ, есенціальну гіпертензію) і симптоматичні АГ, у тому числі ізольовану (систолічну) АГ.
Термін «гіпертонічна хвороба» вживається тоді, коли етіологія підвищення АТ не з’ясована. Якщо підвищення АТ є проявом якогось конкретного захворювання, воно трактується як симптоматична (вторинна) АГ. Найчастіше це спостерігається при ураженні нирок (пієлонефрит, гломерулонефрит), ендокринної системи (феохромоцитома, хвороба Іценка - Кушинга, синдром Конна та ін), природжених або набутих ураженнях судин (коарктація дуги аорти, звуження судин нирок), нервової системи (пухлини, травми головного мозку).
Етіологія і патогенез. Основним етіологічним фактором ГХ вважають нервовопсихічне перенавантаження ЦНС, спричинене короткочасними гострими або тривалими нервовими негативними впливами. Внаслідок цього виникає активація генетичних дефектів людини. До них належать: порушення мембран клітин; скупчення іонів Са++; активність симпатичної нервової системи; ренінангіотензивна система; рецептори до ангіотензину ІІ; гіпертрофія серця. Всі ці явища регулюються генами і деякою мірою зовнішніми факторами. Внаслідок цього зростають серцевий викид, загальний периферичний судинний опір, розвивається АГ. У свою чергу, АГ спричинює шемію мозку, серця, судин, нирок, надниркових залоз. Експериментально та клінічно доведено, що в цих органах відбувається збільшення вмісту тканинного ангіотензину. Існують різні шляхи утворення ангіотензину ІІ, а не тільки за участю ангіотензинперетворюючого ферменту (АПФ). Циркулюючий ангіотензин ІІ підвищує загальний периферичний судинний опір, що збільшує АТ, а тканинний ангіотензин ІІ збільшує ішемію органів. Тканинний ангіотензин ІІ спричинює гіпертонічну енцефалопатію, порушення мозкового кровообігу, гіпертрофію і фіброз міокарда, гіпертрофію артерій м’язового типу, артеріосклероз, гломерулосклероз, підвищення рівнів катехоламінів і альдостерону. Все це сполучене з добовою динамікою АТ.
Таким чином, ГХ - це полігенний дефект, який поки що не можна подолати. Вилікувати хворих на ГХ неможливо, але активно запобігти її тяжким органним ускладненням - це реально.
Відповідно до останніх рекомендацій ВООЗ та Міжнародного товариства гіпертензії (1999) виділяють декілька рівнів АТ .
Цю класифікацію рекомендується застосовувати для з’ясування стадії як гіпертонічної хвороби (есенціальної гіпертензії), так і вторинної гіпертензії.
Якщо виявлено ГХ, діагноз формулюється з визначенням її стадії та характеру ураження органівмшеней (серце, головний мозок, очне дно, нирки, судини).
Перебіг захворювання відображають стадії підвищення АТ і стан органівмішеней. На початку захворювання самопочуття хворих може бути цілком задовільним або проявлятися у вигляді специфічних церебральних і кардіальних скарг: головний біль у потиличній, тім’яній, скроневій ділянках; запаморочення; шум у голові, вухах; минущі порушення зору; дратівливість; безсоння; колючі болі у ділянці серця, серцебиття, задишка. У міру прогресування хвороби виникають об’єктивні ознаки порушення функцій органівмішеней і різноманітні ускладнення.
Перебіг ГХ часто ускладнюється гіпертонічними кризами. Криз - це гостре погіршання стану хворого, що призводить до значного і відносно короткочасного підвищення АТ. Існують 2 типи кризів. Для кризів І типу характерні нейровегетативні зрушення, які супроводжуються гіперсимпатикотонією, що призводить до різкого збільшення викиду крові за одиницю часу і підвищення загального периферичного судинного опору. Для кризів ІІ типу характерними є підвищення у крові альдостерону, затримка натрію в організмі, що спричинює збільшення об’єму крові.
Гіпертонічний криз І типу супроводжується різким підвищенням АТ, головним чином систолічного, інтенсивним головним болем, запамороченням, пітливістю, тремором, тахікардією, іноді порушенням зору, нудотою, блювотою. Продовжується нетривалий час (від кількох годин до 1 доби) і може закінчитися urina spastica.
Розвиток кризів ІІ типу поступовий, протягом декількох діб, при цьому підвищується як систолічний, так і діастолічний АТ. Хворі на вигляд часто бліді, пригнічені, сонливі; з одутлим обличчям; спостерігаються набряки рук і ніг; у літніх хворих знижене сприйняття кольору.
Гіпертензія малого кола кровообігу (первинна, вторинна). Причини та механізми розвитку. Клінічні та гемодинамічні прояви.
29.20. Які причини і механізми розвитку гіпертензії малого кола кровообігу?
Гіпертензія малого кола кровообігу характеризується збільшенням тиску в легеневій артерії понад 25 мм рт. ст. її розвиток може бути обумовлений такими механізмами:
а) тривалий спазм артеріол легень. Найчастіше виникає в результаті зменшення парціального тиску кисню в альвеолярному повітрі, що буває при гіпоксичній гіпоксії (див. розд. 19) і порушеннях вентиляції легень;
б) гострий рефлекторний спазм легеневих артеріол. Розвивається при емболії судин легень, стенозі отвору двостулкового клапана. В останньому випадку вмикається рефлекс Китаєва: збільшення тиску в лівому передсерді і легеневих венах викликає збудження барорецепторів і спазм легеневих артеріол, що попереджає збільшення гідростатичного тиску в капілярах легень і розвиток набряку;
в) збільшення тиску повітря в бронхах і альвеолах. Викликає здавлення легеневих капілярів і, як наслідок, збільшення судинного опору в малому колі кровообігу. Буває у людей під час важких нападів кашлю. При цьому тиск у легеневій артерії може зростати до 250 мм рт. ст.;
г) облітерація легеневих судин (артеріол, капілярів, венул) унаслідок ураження їхніх стінок (наприклад, при емфіземі легень). В експерименті показано, що гіпертензія малого кола кровообігу виникає при вимиканні не менше 2/3 судинного русла. Отже, видалення однієї легені не призводить до розвитку цього синдрому;
ґ) збільшення хвилинного об'єму серця більше ніж у 3 рази;
д) порушення відтоку крові по легеневих венозних судинах (вади двостулкового клапана серця, недостатність лівого шлуночка, здавлювання легеневих вен);
є) збільшення в 'язкості крові (наприклад, при поліцитемії);
є) уроджені вади, пов'язані зі скиданням крові зліва направо (незарощення Боталло-вої протоки, дефекти міжшлуночкової перегородки). Залежно від того, на якій ділянці легеневих судин збільшується опір, розрізняють пре- і посткапілярну форму гіпертензії малого кола кровообігу.
29.21. Які механізми можуть лежати в основі розвитку набряку
легень?
Вихід рідини з кровоносних судин в інтерстиціальну тканину легень і альвеоли
може бути обумовлений такими механізмами.
1. Гідростатичний механізм - різке збільшення гідростатичного тиску в капілярах легень. Набряк розвивається, коли гідростатичний тиск стає вищим за 30 мм рт. ст. (у нормі 6-9 мм рт. ст.), тобто його величина стає більшою за онкотичний тиск крові. Така ситуація може виникати при гострій лівошлуночковій недостатності серця, обумовленій великим інфарктом міокарда, при стенозі отвору двостулкового клапана (кардіогенний механізм), при введенні великих кількостей (кілька літрів) крово- і плазмозамінних розчинів хворим з порушеним діурезом (гіперво-лемічний механізм).
2. Мембраногенний механізм — збільшення проникності легеневих капілярів. Буває при:
а) екзогенній інтоксикації (отруєння фосфорорганічними сполуками, наприклад, фосгеном);
б) ендогенній інтоксикації (уремія, печінкова недостатність);
в) алергічних реакціях І типу.
3. Онкотичний механізм — зменшення онкотичного тиску плазми крові. Відносно часто буває у хворих з нефротичними синдромом (див. розд. 32).
З урахуванням патогенезу розрізняють дві фази розвитку набряку легень.
I. Інтерстиціальний набряк - накопичення набрякової рідини в інтерстиціальній тканині легень. Клінічно виявляється нападами серцевої астми. Розвивається паренхіматозна недостатність дихання з явищами гіпоксемії.
II. Альвеолярний набряк — перехід набрякової рідини в альвеоли. При цьому порушується їх вентиляція — розвивається вентиляційна недостатність дихання з явищами гіпоксемії і гіперкапнії.
29.22. Якими порушеннями виявляє себе синдром емболії малого кола кровообігу?
Поява емболів у судинах легень викликає розвиток таких змін:
1) генеролізованш спазм артеріол усього малого кола кровообігу (а не тільки судин, де містяться емболи). Це викликає різку гіпертензію малого кола і розвиток гострої правопшуночкової недостатності серця (синдром гострого легеневого серця);
2) зменшення артеріального тиску у великому колі кровообігу. Пов'язане зі зменшенням хвилинного об'єму серця і зниженням тонусу артеріол у периферичних відділах (рефлекс Швічки—Паріна);
3) порушення зовнішнього дихання — розвиток паренхіматозної дихальної недостатності (див. запит. 29.17).
Артеріальна гіпотензія. Етіологія та патогенез гострих і хронічних артеріальних гіпотензій.
28.29. Як класифікують артеріальну гіпотензію? Які гемодинамічні фактори можуть лежати в основі її розвитку?
Артеріальна гіпотензія (стійке зниження артеріального тиску) спостерігається частіше в осіб астенічної конституції і виявляється загальною адинамією, швидкою стомлюваністю, тахікардією, задишкою, запамороченням, головним болем, непритом-ностями і депресивним станом з періодичним підвищенням нервової збудливості.
Артеріальну гіпотензію класифікують у такий спосіб.
I. Фізіологічна (не супроводжується хворобливими симптомами).
II. Патологічна (з характерним симптомокомплексом):
1. Гостра.
2. Хронічна:
а) симптоматична (вторинна);
б) нейроциркуляторна дистонія гіпотензивного типу (первинна).
З огляду на те, що рівень артеріального тиску визначається величиною серцевого виштовху, об'ємом циркулюючої крові і тонусом резистивних судин, можливі три гемодинамічні форми артеріальної гіпотензії:
1) пов'язана з недостатністю скорочувальної функції серця;
2) викликана зменшенням об'єму циркулюючої крові;
3) така, що виникає внаслідок зниження тонусу резистивних судин.
28.30. Які причини і механізми розвитку хронічної артеріальної гіпотензії?
Симптоматична (вторинна) хронічна артеріальна гіпотензія є наслідком низки загальних соматичних гострих і хронічних захворювань серця (вади, міокардит, інфаркт міокарда), головного мозку (комоція), легень (крупозна пневмонія), печінки (гепатит, механічна жовтяниця), крові (анемія), ендокринних залоз, а також екзогенних інтоксикацій.
Стосовно нейроциркуляторної (первинної) артеріальної гіпотензії вважають, що її основним етіологічним і патогенетичним фактором, як і гіпертонічної хвороби, є перенапруження основних процесів кори великого мозку (збудження і гальмування). Однак, на відміну від первинної гіпертензії, спостерігається переважання гальмування і поширення його на підкіркові вегетативні утворення, зокрема на судиноруховий центр.
28.31. Які загальні і місцеві розлади гемодинаміки можуть бути пов'язані з первинними порушеннями функції ємнісних судин?
До ємнісних судин належать вени, що депонують кров з метою її розподілу і повернення до серця. Було відзначено, що в судинах ділянки низького тиску міститься 70-80 % загального об'єму крові. Про важливе функціональне значення венозного відділу свідчить хоча б той факт, що одномоментне зменшення його ємності всього лише на 3 % подвоює венозне повернення до серця, а при однакових за величиною змінах тиску в артеріальній і венозній системах об'єм останньої змінюється приблизно в ЗО разів більше, ніж артеріальної.
З урахуванням сказаного було зроблено висновок про те, що вже незначні порушення функції з боку ємнісних судин можуть призводити до істотних порушень загальної гемодинаміки. Такі розлади можуть виявлятися двома типами змін:
а) розвитком артеріальної гіпертензії при збільшенні тонусу гладких м'язів венозних судин, унаслідок чого збільшується діастолічний приплив крові до серця;
б) виникненням артеріальної гіпотензії— гострої (колапсу) або хронічної — при швидкому або тривалому збільшенні ємності венозної системи.
Місцеві розлади кровообігу, пов'язані з порушенням функції вен, призводять до розвитку венозної гіперемії (дав. розд. 13).
Артеріосклероз: визначення поняття, класифікація. Характеристика основних форм: атеросклероз (Маршана), медіакальциноз (Менкеберга), артеріолосклероз.
Артеріосклероз являє собою комбінацію чотирьох процесів: інфільтрації, проліферації, дегенерації і склерозування. Різні поєднання цих процесів у різних судинах визначають "мозаїчний" характер артеріосклеротичних уражень.
1. Інфільтрація - проникнення із плазми крові в судинну стінку і відкладення в ній ліпідів, складних вуглеводів і білків.
2. Проліферація — розмноження гладком'язових клітин артеріальної стінки, у результаті чого формуються так звані фіброзні "бляшки", що виступають у просвіт артерій і порушують течію крові в них.
3. Дегенерація— цим терміном позначають ушкодження і загибель клітин судинної стінки, а також розвиток дистрофічних змін, у тому числі кальцинозу.
4. Склерозування — посилене утворення сполучної тканини, що виявляється синтезом її основної інтерстиціальної речовини й волокнистих структур.
Атеросклероз. Етіологія атеросклерозу: фактори ризику, причинні фактори. Сучасні теорії атерогенезу - «запальна» і «рецепторна». Роль спадкових і набутих порушень рецептор-опосередкованого транспорту ліпопротеїнів в атерогенезі.
Сучасне тлумачення атеросклерозу набагато ширше. За визначенням ВООЗ, атеросклероз — це різні поєднання змін інтими артерій, що виявляються у вигляді осередкового відкладення ліпідів, складних сполук вуглеводів, елементів крові і циркулюючих у ній продуктів, утворення сполучної тканини і відкладення кальцію.
28.5. Що таке артеріосклероз Менкеберга?
Описана в 1903 р. Менкебергом на прикладі артерій нижніх кінцівок людини форма артеріосклерозу характеризується ураженням середньої оболонки (медії) артерій еластичного і еластично-м'язового типу і виявляється тріадою ознак: медіанекро-зом, медіакальцинозом і медіасклерозом.
28.6. Дайте порівняльну характеристику атеросклерозу й артеріосклерозу Менкеберга.
Показник |
Атеросклероз |
Артеріосклероз Менкеберга |
1.Уражується |
Інтима |
Медіа |
2. Переважають процеси |
Інфільтрації і проліферації |
Дегенерації і склерозування |
3. Переважно відкладаються |
Ліпіди (холестерол) |
Солі кальцію (розвивається кальциноз) |
4. Прояви: |
Стенозування артерій (утворення «бляшок») |
Зменшення еластичності артеріальної стінки |
5. Наслідки: |
Ішемія органів і тканин |
Аневризми судин (розшарування стінки і розрив) |
28.7. Чим виявляють себе склеротичні зміни кровоносних судин?
Склеротично змінені судини вирізняються підвищеною щільністю й крихкістю. Унаслідок зниження еластичних властивостей вони не в змозі адекватно змінювати свій просвіт залежно від потреби органа або тканини у кровопостачанні.
Спочатку функціональна неповноцінність склеротично змінених судин, а отже, органів і тканин виявляється тільки при підвищенні до них вимог, тобто при збільшенні навантаження. Подальше прогресування атеросклеротичного процесу може призвести до зниження працездатності і у стані спокою.
Сильний ступінь атеросклеротичного процесу, як правило, супроводжується звуженням і навіть повним закриттям просвіту артерій. При повільному склерозу ванні артерій в органах з порушеним кровопостачанням відбуваються атрофічні зміни з поступовим заміщенням функціонально активної паренхіми сполучною тканиною.
Швидке звуження або повне перекриття просвіту артерій часто веде до змертвіння ділянки органа з порушеним кровообігом, тобто до інфаркту. Інфаркт міокарда — найчастіше і найнебезпечніше ускладнення атеросклерозу вінцевих артерій.
28.8. Як в експерименті моделюють атеросклероз?
У 1912 р. М. Анічков і С. Халатов запропонували спосіб моделювання атеросклерозу у кролів шляхом уведення всередину холестеролу (через зонд або домішуючи його до звичайного корму). Виражені атеросклеротичні зміни розвиваються через декілька місяців при щоденному використанні 0,5 г холестеролу на 1 кг маси тіла. Як правило, їх супроводжує підвищення рівня холестеролу в сироватці крові (у 3-5 разів, якщо порівнювати з вихідними величинами), що стало підставою для припущення про провідну патогенетичну роль у розвитку атеросклерозу гіперхолес-теролемії. Цю модель легко відтворюють не тільки у кролів, але і в курей, голубів, мавп, свиней.
У собак і щурів, резистентних до дії холестеролу, атеросклероз відтворюють шляхом комбінованого впливу холестеролу і метилтіоурацилу, що пригнічує функцію щитоподібної залози. Таке поєднання двох факторів (екзогенного і ендогенного)
веде до тривалої і вираженої гіперхолестеролемії. Додавання до їжі вершкового масла і солей жовчних кислот також сприяє розвитку атеросклерозу.
У курей (півнів) експериментальний атеросклероз аорти розвивається після тривалої дії діетилстильбестролу. У цьому випадку атеросклеротичні зміни виявляються на тлі ендогенної гіперхолестеролемії, що виникає внаслідок порушення гормональної регуляції обміну речовин.
28.9. Що таке фактори ризику атеросклерозу? Що до них відносять?
Факторами ризику атеросклерозу називають сукупність внутрішніх і зовнішніх умов, які в багато разів підвищують імовірність розвитку цього захворювання у людини.
Проведені в багатьох країнах світу епідеміологічні дослідження дають можливість виділити цілий ряд факторів ризику атеросклерозу.
1. Вік. Різке збільшення частоти і тяжкості атеросклеротичних уражень судин у зв'язку з віком, особливо помітне після ЗО років, стало підставою для того, щоб деякі дослідники вважали атеросклероз функцією віку і винятково біологічною проблемою (І. Давидовський). Більшість учених, однак, дотримуються думки, що вікові і атеросклеротичні зміни судин — це різні форми артеріосклерозу, особливо на пізніх стадіях їхнього розвитку. При цьому вікові зміни судин сприяють розвитку атеросклербтичних уражень. Зазначений аспект проблеми атеросклерозу знайшов своє відображення в роботах М. М. Горева і очолюваної ним лабораторії Інституту геронтології НАН України.
2. Стать. У віці 40—70 років на атеросклероз та інфаркт міокарда атеросклеротичної природи чоловіки хворіють частіше, ніж жінки (у середньому в 3-4 рази). Після 70 років захворюваність на цю недугу серед чоловіків і жінок приблизно однакова. Зазначені відмінності, мабуть, пов'язані, з одного боку, з нижчим вихідним рівнем холестеролу і тим, що у жінок він міститься в сироватці крові в основному у фракції неатерогенних ліпопротеїдів високої густини, а з другого — з антисклеротичною дією жіночих статевих гормонів.
3. Спадковість. Роль спадкового фактора у виникненні атеросклерозу підтверджують статистичні дані про високу частоту ішемічної хвороби серця в окремих родинах, а також у однояйцевих близнюків. Мова йде про спадкові форми гіперліпо-протеїнемії і спадково обумовлені дефекти метаболізму артеріальної стінки.
4. Надлишкове харчування. Досвід країн з високим життєвим рівнем (США, Швеція, Чехія та ін.) переконливо доводить таку закономірність: що більше потреба в енергії задовольняється за рахунок тваринних жирів і продуктів, які містять хо-лестерол, то вищий вміст холестеролу в крові й відсоток захворюваності на атеросклероз. Навпаки, у країнах, де на частку жирів тваринного походження припадає незначна частина енергетичної цінності добового раціону (близько 10 %), захворюваність на атеросклероз низька (Японія, Китай).
Існує також залежність між захворюваністю на атеросклероз і кількістю споживаного цукру. Якщо додати до цього, що 75-85 % хворих на цукровий діабет хворіють на атеросклероз і вмирають від нього, у 4/5 хворих на атеросклероз установлене зниження толерантності до глюкози, а 1/3 з них перебуває в предіабетичному стані, то певну роль у виникненні атеросклерозу варто відвести надмірному споживанню вуглеводів і порушенню їх утилізації.
5. Стрес. Є спостереження, які свідчать про те, що захворюваність на атеросклероз вища серед людей "стресових професій", тобто професій, що вимагають тривалої і сильної нервової напруги (лікарі, учителі, викладачі, працівники управлінського апарату, льотчики та ін.).
У цілому захворюваність на атеросклероз вища серед міського населення, якщо порівнювати з сільським. Це може пояснюватися тим, що в умовах великого міста людина частіше зазнає нейрогенних стресових впливів.
6. Гіподинамія. Малорухливий спосіб життя, різке зменшення фізичного навантаження (гіподинамія) - ще один важливий фактор атерогенезу. Про це, зокрема, свідчать менша захворюваність на атеросклероз серед працівників фізичної праці і більша- в осіб, робота яких пов'язана з розумовою працею; швидша нормалізація рівня холестеролу в сироватці крові, після надмірного його надходження ззовні, під дією фізичних навантажень.
В експерименті виявлено виражені атеросклеротичні зміни в артеріях кролів після того як вони тривалий час перебували у спеціальних клітках, що значно обмежували рухову активність тварин. Особливу атерогенну небезпеку являє собою поєднання малорухливого способу життя і надлишкового харчування. 1. Інтоксикація. Вплив алкоголю, нікотину, інтоксикація бактеріального походження та інтоксикація, викликана різними хімічними речовинами (фториди, CO, H2S, свинець, бензол, сполуки ртуті), також є факторами, що сприяють розвитку атеросклерозу. Більшість наведених тут інтоксикацій супроводжувалася не тільки загальними порушеннями жирового обміну, властивими атеросклерозу, але й типовими дистрофічними та інфільтративно-проліферативними змінами в артеріальній стінці.
8. Артеріальна гіпертензія. Підвищений артеріальний тиск набуває значення фактора, що сприяє розвитку атеросклерозу в комбінації з іншими, особливо якщо він перевищує 160/90 мм рт. ст. Так, при однаковому рівні холестеролу захворюваність на інфаркт міокарда при гіпертензії в п'ять разів вища, ніж при нормальному артеріальному тиску. В експерименті на кролях, у їжу яких додавали холестерол, атеросклеротичні зміни розвиваються швидше і досягають більшого ступеня на тлі артеріальної гіпертензії.
9. Гормональні порушення, хвороби обміну речовин. У деяких випадках атеросклероз виникає на тлі попередніх гормональних порушень (цукровий діабет, мікседема, зниження функції статевих залоз) або хвороб обміну речовин (подагра, ожиріння, спадкові форми гіперліпопротеїнемії і гіперхолестеролемії). Про етіологічну роль гормональних розладів у розвитку атеросклерозу свідчать і досліди з експериментального відтворення цих порушень у тварин шляхом впливу на ендокринні залози.
28.10. Які існують концепції патогенезу атеросклерозу?
Відомі нині теорії патогенезу атеросклерозу можна звести до двох, принципово різних концепцій, що відрізняються між собою відповіддю на питання: що первинне, а що вторинне при атеросклерозі, інакше кажучи, що є причиною, а що наслідком - ліпоїдоз внутрішньої оболонки артерій чи дегенеративно-проліферативні зміни останньої.
Відповідно до уявлень R Вірхова і його послідовників, при атеросклерозі спочатку розвиваються дистрофічні зміни внутрішньої оболонки стінки артерій, а відкладення ліпідів і солей кальцію — явище вторинного порядку. Перевагою даної концепції є те, що вона спроможна пояснити розвиток спонтанного і експериментального атеросклерозу як у тих випадках, коли є порушення ліпідного обміну, так і в тих (що особливо важливо), коли їх немає. Першорядну роль автори цієї концепції відводять артеріальній стінці, тобто субстрату, який безпосередньо втягується в патологічний процес.
На противагу цим поглядам ще відтоді, коли М. Анічков і С. Халатов провели перші свої експерименти, успішно розвивається концепція про роль у розвитку атеросклерозу загальних метаболічних порушень в організмі, що супроводжуються гі-перхолестеролемією і гіперліпопротеїнемією. З цих позицій атеросклероз — наслідок первинної дифузної інфільтрації ліпідів, зокрема холестеролу, у незмінену внутрішню оболонку артерій. Подальші зміни в судинній стінці (явища мукоїдного набухання, дистрофічні зміни волокнистих структур і клітинних елементів субендотеліаль-ного шару, продуктивні зміни) розвиваються у зв'язку з відкладенням у ній ліпідів, тобто є вторинними.
28.11. У чому сутність плазмової теорії патогенезу атеросклерозу?
Відповідно до плазмової теорії, основу патогенезу атеросклерозу та інфільтрації судинної стінки зокрема, становлять зміни хімічного складу плазми крові, що виникають унаслідок загальних порушень ліпідного обміну в організмі.
Пріоритет у становленні цієї теорії належить М. М. Ані-чкову та його послідовникам.
Плазмова теорія у своєму розвитку пройшла два етапи. ^
Перший етап — холестероловий. Сутність теорії на цьому етапі її розвитку зводилася до положення: "без холестеро-лу немає атеросклерозу". Вважалося, що причиною виникнення інфільтративних змін артеріальної стінки (ліпоїдозу) є збільшення вмісту холестеролу в плазмі крові — гіперхолес-теролемія.
Після того, як стало відомо, що транспорт ліпідів, у тому числі і холестеролу, здійснюється у складі лшопротеїдів, виявилося, що для розвитку атеросклерозу має значення не стільки гіперхолестеролемія, скільки кількісні і якісні зміни ліпо-~~ протеїдів плазми крові. Настав другий етап розвитку плазмової теорії —ліпопротеїновий. Основою його стало положення: "без атерогенних ліпопротеїдів немає атеросклерозу
Недостатність зовнішнього дихання: визначення поняття, принципи класифікації. Патогенез основних клінічних проявів. Задишка: види, причини, механізми розвитку. Хвороба гіалінових мембран. Характеристика.
Недостатність зовнішнього дихання — це патологічний стан, при якому система зовнішнього дихання не здатна забезпечити нормальний склад газів крові (газовий гомеостаз).
29.2. Як класифікують дихальну недостатність ?
I. За клінічним перебігом розрізняють гостру і хронічну недостатність дихання. Гостра недостатність розвивається протягом кількох днів, годин і навіть хвилин, її прикладом може бути асфіксія (див. запит. 29.16).
Хронічна недостатність розвивається протягом тривалого часу і є наслідком захворювань бронхів і легень (хронічна пневмонія, пневмосклероз, емфізема легень та ін.).
II. За вираженістю клінічних ознак недостатність дихання може бути компенсованою і декомпенсованою.
При компенсованій недостатності газовий склад крові ще не змінений (спрацьовують компенсаторні захисні механізми); при декомпенсованій - газовий гомеостаз порушений.
III. За патогенезом виділяють два різновиди: а) вентиляційну і б) паренхіматозну недостатність зовнішнього дихання.
Недостатність зовнішнього дихання - патологічний стан, при якому система зовнішнього дихання не здатна забезпечити нормальний склад газів крові (газовий гомеостаз).
• Дихальну недостатність класифікують:
1) За клінічним перебігом розрізняють: а) гостру і б) хронічну недостатність дихання.
Гостра недостатність розвивається протягом декількох днів, годин і навіть хвилин (асфіксія). Хронічна недостатність розвивається протягом тривалого часу і є наслідком захворювання бронхів і легень (хронічна пневмонія, пневмосклероз, емфізема легень і ін).
2) За клінічними проявами недостатність дихання може бути: а) компенсованою і б) декомпенсованою.
При компенсованій недостатності газовий склад крові не змінюється із-за включення компенсаторних захисних механізмів; при декомпенсованій - газовий гомеостаз порушується.
3) За патогенезом виділяють два різновиди: а) вентиляційну і б) паренхіматозну.
• Вентиляційна недостатність дихання виникає внаслідок порушень обміну газів між атмосферним повітрям і альвеолами легень, тобто в результаті порушень легеневої вентиляції (гіповентиляції).
Для цього виду дихальної недостатності характерні наступні порушення гомеостазу:
1) зменшення напруги кисню (рО2) в артеріальній крові - гіпоксемія;
2) збільшення напруги вуглекислого газу (рСО2) в артеріальній крові - гіперкапнія;
3) газовий ацидоз.
Причини вентиляційної недостатності дихання.
□ Позалегеневі причини - безпосередньо не пов’язані із порушеннями бронхів і легень. Ними можуть бути:
1) порушення функції дихального центру, внаслідок: а) прямого впливу на нього різних патогенних факторів чи б) рефлекторного впливу на хемо- або барорецептори;
2) порушення функції мотонейронів спинного мозку, які іннервують дихальні м’язи, при пухлинах в спинному мозку, сирінгомієлії, поліомієліті;
3) порушення функції нервово-м’язового апарату при: а) ушкодженні нервів, які іннервують дихальні м’язи (запалення, авітаміноз, травма), б) затруднені передачі до м’язів нервового імпульсу (міастенія, ботулізм, правець), в) порушенні функції самих дихальних м’язів (міозит, дистрофія).
Велике значення в акті дихання приймає діафрагма. Порушення роботи діафрагми може привести до значних розладів дихання, що трапляється при ушкодженні n.frenicus. При клонічних судомах м’язів діафрагми з’являється гикавка;
4) порушення рухливості грудної клітки, що веде до обмеження розтягання легень і зменшення альвеолярної вентиляції, при: а) вроджених або набутих деформація ребер і хребетного стовпа, скостенінні реберних хрящів, б) зрощенні листків плеври, в) асциті, метеоризмі, великій тучності, г) різких больових відчуттях, які виникають під час дихання (міжреберна невралгія, міозит);
5) порушення цілісності грудної клітки і плевральної порожнини та попадання у неї атмосферного повітря (пневмоторакс).
□ Легеневі причини - пов’язані із патологічними процесами в легенях і повітряноносних шляхах, зокрема:
1) порушення прохідності повітроносних шляхів (бронхіти, бронхіальна астма, злоякісні пухлини);
2) порушення еластичнких властивостей легеневої тканини (емфізема, пневмосклероз);
3) зменшення кількості функціонуючих альвеол (пневмонія, набряк легень, ателектаз, пневмоторакс).
• Патогенетичні варіанти вентиляційної недостатності дихання включають в себе: 1) дисрегуляційну, 2) рестрикційну та 3) обструкційну недостатність
Дисрегуляторні порушення альвеолярної вентиляції. Причини і механізми патологічного дихання (порушення частоти, глибини, ритму). Патогенез періодичного дихання у дітей.
Дисрегуляційна недостатність виникає під впливом дії рефлекторних, гуморальних або інших чинників на дихальний центр можуть змінюватися ритм, глибина і частота дихання, а також може виникати задишка. Ці зміни можуть бути проявом компенсаторних реакцій організму, спрямованих на підтримку сталості газового складу крові, або проявом порушень нормальної регуляції дихання, що ведуть до розладів альвеолярної вентиляції, до недостатності дихання.
Зміни центральної регуляції дихання можуть виявляти себе такими його типами. 1. Брадипное — рідке дихання. Механізм розвитку рідкого дихання полягає в зміні характеру нервової імпульсації, що йде від різних рецепторів до дихального центру, або в первинному порушенні діяльності самих дихальних нейронів. Рефлекторне зменшення частоти дихання може спостерігатися при підвищенні артеріального тиску (рефлекс із барорецепторів дуги аорти і каротидного синуса),
при гіпероксії (унаслідок періодичного збудження хеморецепторів, чутливих до зниження напруги кисню в артеріальній крові).
Глибоке рідке дихання може з'явитися при підвищенні опору руху повітря у верхніх дихальних шляхах — стенотичне дихання. У цьому випадку вдих і видих відбуваються повільніше, ніж звичайно. У встановленні такого типу дихання певну роль відіграють імпульси, що надходять у дихальний центр від міжреберних м'я-. зів, що працюють з підвищеним навантаженням. Крім того, має значення запізнювання в цьому випадку гальмівного рефлексу Герінга-Брейєра. Брадипное може розвиватися в результаті безпосередньої дії патогенних факторів на дихальний центр, що знижує збудливість дихальних нейронів. Пригнічення дихального центру можливе при тривалій і тяжкій гіпоксії (в умовах розрідженої атмосфери, при недостатності кровообігу та ін.), за умов впливу речовин з наркотичною дією, при деяких органічних ураженнях головного мозку (запалення, порушення мозкового кровообігу, набряк та ін.) і функціональних розладах центральної нервової системи (невроз, істеричні реакції). У всіх цих випадках рідке дихання може супроводжуватися зменшенням його глибини, що призводить до зниження альвеолярної вентиляції і розвитку недостатності дихання.
2. Поліпное (тахіпное) — часте поверхневе дихання. В основі розвитку поліпное лежить рефлекторна перебудова роботи дихального центру. У деяких тварин (наприклад, у собак) часте поверхневе дихання виникає при дії високої температури. У людини поліпное може спостерігатися при гарячці, функціональних порушеннях центральної нервової системи (істерія), ураженні легень (ателектаз, пневмонія, застійні явища).
Крім того, до розвитку поліпное може спричинятися біль, що локалізується у ділянках тіла, які беруть участь у дихальному акті (грудна клітка, черевна стінка, плевра). Біль призводить до обмеження глибини дихання і збільшення його частоти (щадне дихання).
Поліпное знижує ефективність дихання, тому що при цьому значно зменшується альвеолярна вентиляція і газообмін в основному відбувається у "мертвому" просторі.
3. Гіперппое — глибоке часте дихання. В умовах патології гіперпное розвивається при інтенсивній рефлекторній або гуморальній стимуляції дихального центру, наприклад, при зниженні парціального тиску кисню у вдихуваному повітрі або при підвищенні в ньому концентрації С02, при анемії, ацидозі і т. д.
Крайній ступінь збудження дихального центру виявляється диханням Куссмауля, яке найчастіше спостерігається у хворих у стані діабетичної коми. Воно являє собою гучне часте дихання, при якому після глибокого вдиху настає посилений видих з активною участю експіраторних м'язів.
4. Апное— тимчасова зупинка дихання. Тимчасова зупинка дихання може бути пов'язана зі зменшенням рефлекторної або безпосередньої хімічної стимуляції дихального центру. Наприклад, апное виникає у тварини або людини після пасивної гіпервентиляції під наркозом унаслідок зменшення в артеріальній крові напруги CO і припиняється зразу ж, як тільки вміст С02 нормалізується. При швидкому
підвищенні артеріального тиску, наприклад при введенні в кров адреналіну, також, спостерігається апное (рефлекс із барорецепторів). Апное, що часто повторюється і порушує звичайний ритм дихання, може бути пов'язане зі зниженням збудливості нейронів дихального центру (гіпоксія, інтоксикація, органічні ураження головного мозку).
29.7. Що таке періодичне дихання? Які відомі його форми?
Періодичним диханням називають таке порушення ритму дихання, при якому періоди дихання чергуються з періодами апное. Найчастіше бувають два типи періодичного дихання: дихання Чейна-Стокса і дихання Біота.
Дихання Чейна—Стокса характеризується наростанням амплітуди дихання до вираженого гіперпное, а потім зменшенням її до апное, після якого знову настає цикл дихальних рухів, що закінчуються також апное (рис. 135).
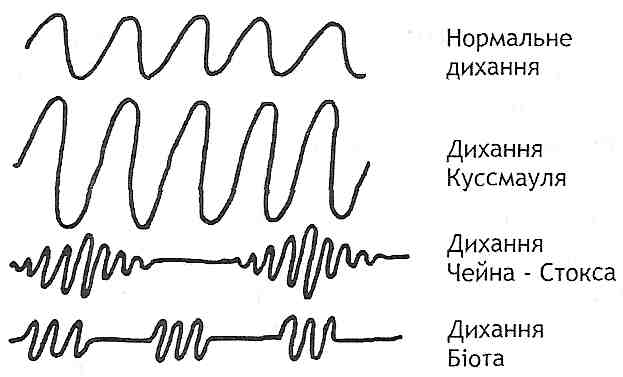
Рис. 135. Дихання Куссмауля і деякі види періодичного дихання
Циклічні зміни дихання у людини можуть супроводжуватися потьмаренням свідомості в період апное і його нормалізацією в період збільшення вентиляції. Артеріальний тиск при цьому також коливається, як правило, підвищуючись у фазі посилення дихання і знижуючись у фазі його ослаблення.
У більшості випадків дихання Чейна-Стокса є ознакою гіпоксії головного мозку. Воно може виникати при недостатності серця, захворюваннях мозку і його оболонок, уремії. Деякі лікарські препарати (наприклад, морфін) також можуть викликати дихання Чейна — Стокса. Його можна спостерігати і у здорових людей на великій висоті (особливо під час сну), у недоношених дітей, що, очевидно, пов'язане з недосконалістю нервових центрів.
Патогенез дихання Чейна-Стокса можна представити в такий спосіб. Клітини кори великого мозку і підкіркових утворень унаслідок гіпоксії пригнічуються — дихання зупиняється, свідомість зникає, пригнічується діяльність судинорухового центру. Однак хеморецептори при цьому все ще здатні реагувати на зміни вмісту газів
у крові. Різкого посилення імігульсації з хеморецепторів, поряд із прямою дією на центри високої концентрації вуглекислого газу і стимулами з барорецепторів унаслідок зниження артеріального тиску, виявляється достатньо для збудження дихального центру — дихання поновлюється. Поновлення дихання веде до оксигенації крові, що зменшує гіпоксію головного мозку і поліпшує функцію нейронів судинорухового центру. Дихання стає глибше, свідомість проясняється, підвищується артеріальний тиск, поліпшується наповнення серця. Вентиляція, що збільшується, веде до підвищення напруги кисню і зниження напруги С02 в артеріальній крові. Це у свою чергу призводить до ослаблення рефлекторної і хімічної стимуляції дихального центру, діяльність якого починає вгасати, - настає апное.
Дихання Біота відрізняється від дихання Чейна-Стокса тим, що дихальні рухи, які характеризуються постійною амплітудою, раптово припиняються, так само як і раптово починаються. Найчастіше дихання Біота спостерігається при менінгіті, енцефаліті та інших захворюваннях, що супроводжуються ушкодженням центральної нервової системи, особливо довгастого мозку.
29.8. Що таке термінальне дихання? Чим воно характеризується?
Термінальним називають дихання, що виникає в термінальних станах, тобто за умов, коли організм перебуває на межі життя і смерті. Найчастіше реєструють два типи термінального дихання: апнейстичне і гаспінг-дихання.
Апнейстичие дихання характеризується конвульсивним зусиллям вдихнути, що не припиняється, але зрідка переривається видихом. В експерименті його спостерігають після перетинання у тварини обох блукаючих нервів і мозкового стовбура між пневмотаксичним (у ростральній частині мосту) і апнейстичним (у середній і каудальній частинах мосту) центрами (рис. 136). Вважають, що апнейстичний центр має здатність збуджувати інспіраторні нейрони, які періодично гальмуються імпульсами із блукаючого нерва і пневмотаксичного центру. Перетинання зазначених структур призводить до постійної інспіраторної активності апнейстичного центру.
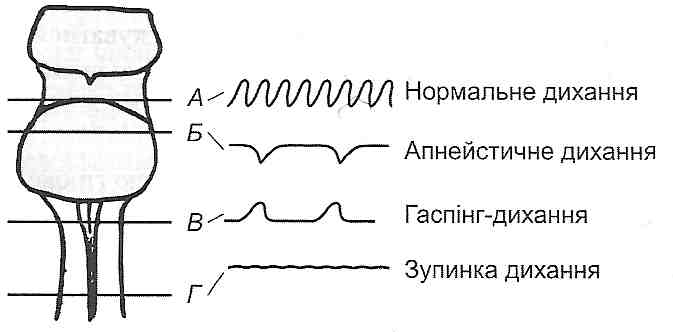
Рис. 136. Розвиток термінального дихання після перетинання стовбура мозку нарізних його рівнях (А, Б, В, Г)
Гаспінг-дихання - це поодинокі, рідкі вдихи, частота і амплітуда яких поступово зменшується аж до повної зупинки дихання. Такий вид дихання спостерігають
при агонії, наприклад у заключній стадії асфіксії. Звичайно характерні для гаспінг-дихання рідкі і низькоамплітуцні вдихи виникають після тимчасової зупинки дихання (претермінальної паузи). Поява їх, можливо, пов'язана зі збудженням клітин, що містяться в каудальній частині довгастого мозку, після вимикання функції вище розташованих відділів мозку.
29.9. Що таке задишка? Які механізми її розвитку?
Задишка (дисппое) - це відчуття нестачі повітря і пов'язана з цим потреба посилити дихання.
Відчуваючи нестачу повітря, людина не тільки мимовільно, але й свідомо збільшує активність дихальних рухів, прагнучи позбутися цього тяжкого відчуття, наявність якого і є найбільш істотною відмінністю диспное від інших видів порушення регуляції дихання (гіперпное, поліпное та ін.). Тому у людини, що втратила свідомість, задишки не буває.
Задишка виникає у тих випадках, коли впливи, що збуджують вдих, є сильнішими за впливи, що його пригнічують, а також у разі підвищення чутливості дихального центру до чинників, які стимулюють дихання. Найважливішими з цих впливів є такі.
1. Збудження рецепторів, що стимулюють центр вдиху, і які активуються при сильному зменшенні об'єму легеневих альвеол (сильнішому, ніж при максимальному видиху). При патології може виникнути постійна імпульсація від цих рецепторів. Наприклад, при застійних явищах у легенях (недостатність серця, пневмонія) переповнені кров'ю судини, що оточують альвеоли, здавлюють їх, ємність альвеол зменшується, що веде до збудження рецепторів спадіння.
2. Збудження рецепторів інтерстиціальної тканини легень (J-рецепторів). Усі патологічні процеси, що ведуть до застійних явищ у легенях (пневмонія, недостатність серця), можуть викликати тривале збудження J-рецепторів і підвищену стимуляцію дихальних нейронів.
3. Рефлекси з повітроносних шляхів. Можуть викликатися твердими частинками, парами хімічних сполук та іншими чинниками, що потрапили в повітроносні шляхи і подразнюють так звані іритантнірецептори (мають властивості одночасно ме-хано- і хеморецепторів), розташовані в субепітеліальному шарі трахеї, бронхів і бронхіол. Значне збудження іритантних рецепторів спостерігають при бронхітах, бронхопневмонії, бронхіальній астмі і хворобах, при яких у бронхах і альвеолах міститься слиз, ексудат або транссудат).
4. Рефлекси з барорецепторів аорти і сонної пазухи. Ці рефлекси долучаються до патогенезу задишки при крововтраті, шоку, колапсі. За умов зменшення артеріального тиску до рівня 70 мм рт. ст. і нижче, різко зменшується імпульсація від цих рецепторів, яка в нормі гальмує центр вдиху (через активацію центру видиху).
\ 5. Рефлекси з хеморецепторів аорти і сонної пазухи. При зниженні в крові напруги 02, підвищенні напруги С02 або збільшенні концентрації іонів водню відбувається посилене збудження рецепторів, розташованих в аортальному і каротидному тільцях, і, як наслідок, — посилене збудження центру вдиху. Цей механізм відіграє важливу роль у розвитку задишки при ацидозі, недостатності дихання, при анемії і т. д.
6. Безпосередня стимуляція нейронів дихального центру. У довгастому мозку є хеморецептори, вибірково чутливі до вуглекислого газу. Сильне збудження цих рецепторів при гіперкапнії також сприяє розвитку задишки.
7. Рефлекси з дихальних м 'язів. Відчуття нестачі кисню може виникнути при надмірному розтягненні міжреберних м'язів і сильному збудженні рецепторів розтягу, імпульсація від яких надходить у вищі відділи головного мозку. Цей механізм діє під час виконання тяжкої фізичної роботи, що потребує значної роботи інспіра-торних м'язів, при зменшенні еластичності легень, звуженні верхніх дихальних шляхів.
8. Стимуляція дихального центру продуктами власного метаболізму. Ідеться про накопичення вуглекислого газу, кислих продуктів обміну і зниження напруги кисню безпосередньо в нервових центрах унаслідок порушення мозкового кровообігу (спазм або тромбоз судин головного мозку, набряк мозку, колапс).
Дихання при задишці, як правило, часте і глибоке. Посилюється як вдих, так і видих, який за умов задишки носить активний характер і відбувається за участю експіраторних м'язів. Однак у деяких випадках може переважати або вдих, або видих, тоді ведуть мову про інспіраторну (посилений вдих) або експіраторну (посилений видих) задишку. Інспіраторну задишку спостерігають, наприклад, у першій стадії асфіксії, при загальному збудженні центральної нервової системи, при фізичному навантаженні у хворих з недостатністю кровообігу, при пневмотораксі. Експіраторна задишка буває рідше і виникає головним чином при бронхіальній астмі, емфіземі, коли при видиху збільшується опір потокові повітря в нижніх дихальних шляхах
Порушення альвеолярної вентиляції. Обструктивні та рестриктивні механізми розвитку.
29.10. Що таке рестриктивна недостатність дихання? Які її причини?
PecmpuKmuenoio називають дихальну недостатність, пов'язану з обмеженням зовнішнього дихання (restrict - обмеження; рис. 137).
Рис. 137. Зміни показників вентиляції легень при рестриктивній дихальній недостатності (ДН)
Таке обмеження може бути обумовлене: 1) зменшенням дихальної поверхні легень, що буває після видалення сегмента, частки або цілої легені; при руйнуванні великих ділянок легень (туберкульоз); у результаті спадіння легеневої тканини (ателектаз, пневмоторакс);
2) збільшенням пружного опору легень — порушенням їхньої здатності розправлятися під час вдиху. Така ситуація закономірно виникає при:
а) зменшенні розтяжності легень у зв'язку із заміщенням еластичних структур легеневої тканини колагеновими (пневмосклероз);
б) збільшенні сили поверхневого натягу в альвеолах при порушеннях сурфак-танту (зменшенні утворення або посиленому руйнуванні).
29.13. Що таке обструктивна недостатність дихання? Коли вона розвивається?
Обструктивною називають дихальну недостатність, яка виникає внаслідок збільшення аеродинамічного опору повітроносних шляхів. Основним фактором, що викликає таке збільшення, є зменшення радіуса повітроносних трубок (бронхів, бронхіол).
Причинами обструктивної недостатності дихання є:
1) спазм гладких м 'язів бронхів (бронхіальна астма);
2) набряк слизових оболонок (бронхіти, бронхіоліти); м
3) здавлювання бронхіол (емфізема легень).
Бронхіальна астма. Етіологія: фактори ризику, причинні фактори. Патогенез. Клінічна картина. Особливості у дітей.
Бронхіальна астма — це алергічне захворювання, що характеризується повторними нападами експіраторної задишки, викликаної дифузним порушенням прохідності бронхів.
Під час тривалих нападів бронхіальної астми розвивається обструктивна недостатність дихання.
Для бронхіальної астми характерні такі зміни показників зовнішнього дихання (рис. 139): 1) зменшення резервного об'єму видиху; 2) зменшення життєвої ємності
легень; 3) збільшення залишкового об'єму легень; 4) зменшення об'єму форсованого видиху (проба Тифно; рис. 140).
Рис. 139. Зміни показників вентиляції легень при бронхіальній астмі
Рис. 140. Об'єм форсованого видиху (ОФВ; тест Тифно) при обструктивній дихальній недостатності (ДН)
Причини і механізми порушень дифузії газів у легенях. Порушення легеневого кровообігу. Порушення загальних і регіональних вентиляційно-перфузійних взаємовідношень у легенях.
29.18. Назвіть причини порушення дифузії газів у легенях.
Дифузія газів через альвеоло-капілярну мембрану здійснюється відповідно до першого закону Фіка:
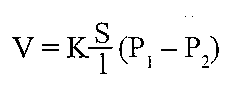
де V - кількість газу, що дифундує за одиницю часу; k - коефіцієнт дифузії; S -загальна площа, через яку відбувається дифузія; 1 - товщина мембрани; Р, і Р2 — парціальний тиск газів по обидві сторони мембрани.
З урахуванням цього можна виділити такі причини порушень дифузії газів у легенях:
1) зменшення коефіцієнта дифузії. Величина його залежить як від природи газу, так і від середовища, у якому відбувається дифузія. Практично має значення зменшення коефіцієнта дифузії кисню у зв'язку зі зміною властивостей легеневої тканини. При цьому перехід С02 із крові в альвеоли, як правило, не міняється, оскільки коефіцієнт його дифузії дуже високий (у 20-25 разів вищий, ніж кисню);
2) зменшення площі дифузії. Має місце при зменшенні дихальної поверхні легень;
3) збільшення товщини альвеоло-капілярноїмембрани;
4) зменшення різниці між парціальним тиском газів в альвеолярному повітрі та їх напругою в крові легеневих капілярів. Така ситуація виникає при всіх порушеннях вентиляції легень;
5) зменшення часу контакту крові з альвеолярним повітрям. Дифузія кисню порушується в тому випадку, якщо час контакту стає менше 0,3 с.
29.19. Назвіть причини порушень легеневої пєрфузії.
Порушення кровообігу в легенях (легеневої пєрфузії) можуть бути зумовлені такими причинами:
а) зменшенням тиску в правому шлуночку (недостатність правого серця, зменшення венозного повернення при крововтраті, шоку, колапсі);
6) збільшенням тиску в лівому передсерді (стеноз отвору двостулкового клапана, лі-вошлуночкова недостатність серця);
в) збільшенням опору судин малого кола кровообігу. Останнє може бути обумовлено рефлекторним збільшенням тонусу артеріол легень, збільшенням в'язкості крові, наявністю перешкод для руху крові (тромбоз, емболія).
29.20. Які причини і механізми розвитку гіпертензії малого кола кровообігу?
Гіпертензія малого кола кровообігу характеризується збільшенням тиску в легеневій артерії понад 25 мм рт. ст. її розвиток може бути обумовлений такими механізмами:
а) тривалий спазм артеріол легень. Найчастіше виникає в результаті зменшення парціального тиску кисню в альвеолярному повітрі, що буває при гіпоксичній гіпоксії (див. розд. 19) і порушеннях вентиляції легень;
б) гострий рефлекторний спазм легеневих артеріол. Розвивається при емболії судин легень, стенозі отвору двостулкового клапана. В останньому випадку вмикається рефлекс Китаєва: збільшення тиску в лівому передсерді і легеневих венах викликає збудження барорецепторів і спазм легеневих артеріол, що попереджає збільшення гідростатичного тиску в капілярах легень і розвиток набряку;
в) збільшення тиску повітря в бронхах і альвеолах. Викликає здавлення легеневих капілярів і, як наслідок, збільшення судинного опору в малому колі кровообігу. Буває у людей під час важких нападів кашлю. При цьому тиск у легеневій артерії може зростати до 250 мм рт. ст.;
г) облітерація легеневих судин (артеріол, капілярів, венул) унаслідок ураження їхніх стінок (наприклад, при емфіземі легень). В експерименті показано, що гіпертензія малого кола кровообігу виникає при вимиканні не менше 2/3 судинного русла. Отже, видалення однієї легені не призводить до розвитку цього синдрому;
ґ) збільшення хвилинного об'єму серця більше ніж у 3 рази;
д) порушення відтоку крові по легеневих венозних судинах (вади двостулкового клапана серця, недостатність лівого шлуночка, здавлювання легеневих вен);
є) збільшення в 'язкості крові (наприклад, при поліцитемії);
є) уроджені вади, пов'язані зі скиданням крові зліва направо (незарощення Боталло-вої протоки, дефекти міжшлуночкової перегородки). Залежно від того, на якій ділянці легеневих судин збільшується опір, розрізняють пре- і посткапілярну форму гіпертензії малого кола кровообігу.
29.23. Які зміни загальних і місцевих вентиляційно-перфузійних відношень можуть спричинятися до розвитку недостатності зовнішнього дихання?
Вешпшяційно-перфузійне відношення для легень у цілому визначається як відношення хвилинного об'єму альвеолярної вентиляції (Va) до хвилинного об'єму крові (Q). У нормі:
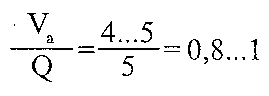
Якщо зазначене відношення більше одиниці, то це свідчить про збільшення функціонального мертвого простору, унаслідок чого ефективність вентиляції погіршується. Така ситуація виникає при гіпервентиляції, не підкріпленій збільшенням перфузії, або при нормальній вентиляції, але порушеному легеневому кровообігу.
Вентиляційно-перфузійне відношення менше 0,8 свідчить про так званий ефект шунтування, коли кров, не збагачена киснем, потрапляє в легеневі вени і потім у велике коло кровообігу. Це буває, коли величина легеневої перфузії значно перевищує величину альвеолярної вентиляції.
В умовах патології у зв'язку з нерівномірністю вентиляції і перфузії різних альвеол вентиляційно-перфузійні відношення в різних ділянках легень можуть бути різними. При цьому вентиляційно-перфузійне відношення для легень у цілому може бути в нормі, хоча і розвиваються ознаки дихальної недостатності. Вони обумовлені існуванням в уражених легенях трьох типів альвеол (рис. 141):
Рис. 141. Місцеві вентиляційно-перфузійні відношення:
1 — ефективний альвеолярний об 'єм;
2 — альвеолярний мертвий простір;
3 — альвеолярний артеріо-венозний шунт
1) альвеоли, які оптимально вентилюються і перфузуються (Va/Q = 0,8... 1). Вони утворюють ефективний альвеолярний об 'єм і становлять більшість альвеол здорових легень;
2) альвеоли, які вентилюються, але не перфузуються (V7Q >1). їхня сукупність становить альвеолярний мертвий простір^
3) альвеоли, які перфузуються, але не вентилюються (V/Q <0,8). З ними пов'язане артеріовенозне шунтування.
Збільшення кількості альвеол другого і третього типів може призводити до розвитку гіпоксемії. При цьому виділення С02 не порушується завдяки високому коефіцієнту його дифузії (розвивається паренхіматозна недостатність дихання).
Асфіксія: визначення поняття, причини, патогенез. Термінальне дихання.
Асфіксія (ядуха) - це загрозливий для життя стан, при якому гостра дихальна недостатність досягає такого ступеня, що у кров зовсім не надходить кисень, а з крові не виводиться вуглекислий газ.
Найчастіше асфіксія настає у разі здавлення дихальних шляхів (задушення), закупорки їхнього просвіту (сторонні предмети, запальний набряк), наявності рідини в дихальних шляхах і альвеолах (утоплення, набряк легень, потрапляння блювотних мас), у разі двостороннього пневмотораксу. Крім того, асфіксія може розвитися при сильному пригніченні дихального центру, порушенні проведення нервових імпульсів до дихальних м'язів, різкому обмеженні рухомості грудної клітки.
У перебігу асфіксії виділяють три періоди.
Перший період асфіксії характеризується швидким збільшенням глибини і частоти дихання з переважанням фази вдиху над фазою видиху. Розвивається загальне збудження, підвищується тонус симпатичної частини вегетативної нервової систе-
ми (розширюються зіниці, з'являється тахікардія, підвищується артеріальний тиск), можливі судоми.
У другому періоді частота дихання поступово зменшується при збереженій максимальній амплітуді дихальних рухів, посилюється фаза видиху. Переважає тонус парасимпатичної частини вегетативної нервової системи (зіниці звужуються, артеріальний тиск знижується, відзначається брадикардія).
В третьому періоді асфіксії спостерігають зменшення амплітуди дихання, його частоти і, нарешті, зупинку дихання. Артеріальний тиск значно знижується. Після короткочасної зупинки дихання, як правило, з'являється кілька рідких конвульсивних дихальних рухів (гаспінг-дихання), після яких настає параліч дихання.
Явища, що їх спостерігають при асфіксії, пов'язані спочатку з накопиченням в організмі вуглекислого газу. Діючи рефлекторно і безпосередньо на дихальний центр, С02 збуджує його, доводячи глибину і частоту дихання до максимально можливих величин. Крім того, дихання рефлекторно стимулюється і зниженням у крові напруги кисню. У міру збільшення в крові вмісту С02 підвищується і артеріальний тиск. Експерименти з вдиханням газових сумішей, що містять 10-20 % С02, показали, що таке підвищення пов'язане, по-перше, з рефлекторним впливом хеморецепторів на судиноруховий центр, по-друге, з посиленим викидом адреналіну в кров, по-третє, зі збільшенням хвилинного об'єму крові в результаті підвищення тонусу вен і збільшення надходження крові до серця при посиленні дихання.
При подальшому збільшенні концентрації С02 у крові починає виявлятися його наркотична дія, рН крові знижується до 6,8-6,5. Посилюється гіпоксемія і, відповідно, гіпоксія головного мозку. Це призводить, у свою чергу, до пригнічення дихання, зниження артеріального тиску. В результаті настає параліч дихання і зупинка серця.
Причини і механізми порушення травлення в порожнині рота. Етіологія, патогенез, експериментальні моделі карієсу та пародонтозу. Причини, механізми порушень слиновиділення.
Недостатність травлення - це патологічний стан, при якому травна система не забезпечує засвоєння поживних речовин, що надходять в організм. Наслідком цього є розвиток різного ступеня голодування (див. розд. 18).
30.2. Як класифікують недостатність травлення?
I. За клінічним перебігом виділяють гостру і хронічну недостатність травлення.
II. Відповідно до анатомічного принципу недостатність травлення може бути обумовлена порушеннями цього процесу:
а) у порожнині рота;
б) у шлунку;
в) у кишках.
III. Недостатність травлення може бути загальною (тотальною) і селективною (парціальною). При загальній недостатності порушено засвоєння всіх поживних речовин, при селективній - тільки окремих їхніх класів (наприклад, ліпідів, лактози, вітаміну В12 та ін.).
IV. За етіологією розрізняють спадково обумовлену (деякі види мальабсорбції) і набуту недостатність травлення. Остання може бути:
а) інфекційного походження;
б) обумовленою впливами фізичних факторів (наприклад, при гострій променевій хворобі);
в) пов'язаною із впливами хімічних агентів;
г) диерегуляторною; ґ) аліментарною.
V. Патофізіологічний принцип передбачає поділ недостатності травлення на три варіанти. Цс недостатність, обумовлена порушеннями:
а) рухової функції травної системи;
б) секреторної її функції;
в) процесів усмоктування.
30.21. Що може спричинятися до порушень жування? Яке значення для травлення мають ці порушення?
Причини порушень жування:
1) ураження зубів та їх відсутність (карієс, пародонтит);
2) ураження жувальних м'язів (міозит);
3) порушення іннервації жувальних м'язів (бульбарні паралічі, неврити);
4) ураження скронево-нижньощелепних суглобів;
5) травматичне ушкодження кісток (нижньої, верхньої щелепи);
6) ураження слизової оболонки порожнини рота і ясен (стоматит, гінгівіт);
7) ураження м'язів і слизової язика;
8) гіпосалівація.
Порушення жування мають такі наслідки:
а) зменшення рефлекторної секреції шлункового і підшлункового соків;
б) уповільнення травлення в шлунку;
в) травматизація слизової оболонки порожнини рота, стравоходу, шлунку;
г) відмова від споживання деяких продуктів харчування, що потрібні організму, але потребують пережовування.
30.22. Що таке карієс зубів? Які причини його розвитку?
Карієс зубів - це патологічний процес, що виявляє себе демінералізацією і про-гресуючим руйнуванням твердих тканин зуба з утворенням дефектів у вигляді порожнин.
Поширеність карієсу становить 80-90 %, а в деяких районах земної кулі - 100 %.
Етіологію карієсу нині остаточно не встановлено, хоча й доведено значення цілого ряду факторів у його розвитку. До таких відносять:
1. Мікроорганізми. Велике значення мають деякі штами стрептококів і лактобакте-рій. В експерименті доведено, що у тварин-гнотобіонтів (безмікробних тварин) карієс не виникає.
2. Надмірне споживання харчових продуктів, що легко ферментуються у порожнині рота бактеріями. Такими, зокрема, є вуглеводи (сахароза, глюкоза, фруктоза^ Запропоновано карієсогенні дієти (містять від 25 до 65 % сахарози), що викликають розвиток карієсу у щурів, хом'яків, мишей.
3. Недостатній вміст у раціоні деяких речовин, зокрема вітамінів групи В, кальцію, фосфору, мікроелементів (фтору, стронцію, молібдену).
4. Генетичні фактори, що впливають на закладку зубних зачатків. Отримано чисті лінії щурів, стійких і чутливих до розвитку карієсу.
5. Зменшення захисної і трофічної функції слини.
6. Загальні порушення фосфорно-кальцієвого обміну (гіпокальціємічні і гіпофосфа-темічні стани; див. розд. 24).
30.23. Як уявляють сьогодні патогенез карієсу?
У патогенезі карієсу провідне значення надають зубному нальоту (зубній бляшці) - скупченню мікроорганізмів та продуктів їх життєдіяльності на поверхні зубів.
Мікроорганізми зубного нальоту виділяють цілий ряд карієсогенних факторів. До них відносять:
а) органічні кислоти (молочна, піровиноградна та ін.), що викликають розчинення оксіапатиту емалі й дентину, внаслідок чого розвивається їх демінералізація;
б) речовини-хелатори (пептиди, амінокислоти). Вони утворюють комплекси з кальцієм, забираючи його від оксіапатитів і викликаючи цим демінералізацію твердих тканин зуба;
в) протеолітичні ферменти. Діючи на білкові компоненти зуба, вони руйнують ма-трикс емалі і дентину. Відбувається деструкція твердих тканин, порушується їх ремінералізація.
Інші етіологічні фактори карієсу (див. запит. 30.22) можуть втручатися в патогенез на різних його етапах. Так, зменшення захисних властивостей слини сприяє розвитку мікрофлори і утворенню зубного нальоту; вуглеводи їжі, адсорбуючись на поверхні зуба, є джерелом великої кількості карієсогенних органічних кислот; генетичні фактори (неповноцінність органічної матриці зуба) і дефіцит цілого ряду речовин сприяють порушенням ремінералізації емалі і дентину. Нарешті, загальні порушення фосфорно-кальцієвого обміну самі собою можуть обумовлювати демінералізацію твердих тканин зуба.
ЗО. 24. Що таке пародонтит? Які фактори можуть лежати в основі його розвитку?
Пародонтит — це дистрофічно-запальне ураження пародонта, тобто тканин, що оточують корінь зуба (періодонта, кісткової тканини зубної комірки, ясен, окістя). Хвороба виявляє себе резорбцією зубних комірок, гноєтечею з ясен, розхитуванням і випадінням зубів.
Серед численних факторів, що мають стосунок до етіології пародонтиту, виділяють такі.
1. Мікрофлора порожнини рота, з якою пов'язують виділення ферментів і біологічно активних речовин зубного нальоту і утворення зубного каменю, що порушує кровопостачання тканин пародонта.
2. Аліментарні порушення — неповноцінне харчування, дефіцит вітамінів, особливо С, Р і Е.
3. Нейрогенні фактори - емоційна перенапруга, що викликає стрес; порушення нервової трофіки.
4. Ендокринні розлади — гіпогонадизм, гіпотиреоз, гіпоінсулінізм, гіперпаратиреоз.
5. Аномалії прикусу, зменшення або надмірне збільшення навантаження на жувальний апарат.
30.25. Які причини і значення гіперсалівації?
Гіперсалівація - збільшення утворення і секреції слини — може бути обумовлена
такими причинами:
а) збудженням рецепторів порожнини рота, стравоходу і шлунку (рефлекторний механізм). Гіперсалівація супроводжує розвиток запальних процесів у порожнині рота (гінгівіти, стоматити, пульпіти, періодонтити), є реакцією надію бормашини;.
б) збудженням центру слиновиділення, що міститься в довгастому мозку (бульбарні паралічі);
в) подразненням вегетативних нервів, що іннервують слинні залози. При стимуляції парасимпатичних нервів виділяється багато рідкої слини, при активації симпатичних - мала кількість, але густої, в'язкої слини;
г) впливом т-холіноміметиків - фармакологічних препаратів, що активують т- хо-лінорецептори;
ґ) впливом гуморальних регуляторних факторів. Цей механізм лежить в основі так званої парадоксальної, або паралітичної, гіперсалівації. Вона починається через добу після повної денервації слинних залоз, досягає максимуму через 6-7 діб, з 15-ї доби починає зменшуватися і через 35-40 діб повністю зникає. У патогенезі парадоксальної гіперсалівації провідне значення має підвищення чутливості денервованих залоз до циркулюючих у крові гуморальних факторів (ацетилхоліну, гістаміну). При гіперсалівації виділяється від 5 до 20 л слини за добу (у нормі - 0,5-2 л). Це
призводить до:
1) зневоднення організму — розвивається гіперосмолярна гіпогідрія, оскільки слина гіпотонічна стосовно крові; ( 2) нейтралізації шлункового соку, що пов'язано зі слабколужним середовищем слини. Ця обставина викликає порушення травлення у шлунку.
30.26. Які причини і значення гіпосалівації?
Причинами зменшення утворення і виділення слини (гіпосалівації) можуть бути: І а) центральне гальмування секреції слинних залоз (страх, переляк, біль); [ б) дія m-холінолітиків — фармакологічних агентів, що блокують т-холінорецептори
периферичних тканин; І в) ушкодження секреторних клітин слинних залоз (запалення, пухлини);
г) порушення виведення секрету (закупорка проток слинних залоз каменями); І г) зневоднення організму. Наслідки гіпосалівації:
1) порушення жування, формування харчової грудки, ковтання, що спричиняється до розладів наступних етапів травлення;
2) травматизація слизової оболонки рота з розвитком її запалення (стоматиту);
3) активний розвиток мікроорганізмів. Він обумовлений зменшенням бактерицидної дії слини (мало лізоциму) і утворенням нальоту зі злущеного епітелію на поверхні язика (прекрасне поживне середовище для мікробів);
4) порушення трофічних впливів слини на зуби, що сприяє розвитку карієсу.
Загальна характеристика порушень моторної і секреторної функцій шлунка. Патологічна шлункова секреція, її типи. Роль нервових та гуморальних механізмів у порушенні секреції.
30.28. Що таке шлункові дискінезії? Які існують їхні варіанти?
Шлунковими дискінезіями називають порушення рухової (моторної) функції шлунку.
Виділяють два варіанти шлункових дискінезій: гіпертонічний і гіпотонічний.
Для гіпертонічного варіанта характерне збільшення тонусу м'язів шлунку (гіпертонія) і посилення його перистальтики (гіперкінезія).
Гіпотонічний варіант, навпаки, характеризується гіпотонією і гіпокінезією.
30.29. Назвіть причини /'значення гіпертонічних дискінезій шлунку.
Причинами рухових розладів шлунку гіпертонічного типу можуть бути:
а) деякі харчові фактори (груба їжа, алкоголь);
б) підвищення шлункової секреції;
в) збільшення тонусу блукаючого нерва;
г) деякі гастроінтестинальні гормони, зокрема мотилін.
Гіпертонія і гіперкінезія шлунку спричинюються до:
1) тривалої затримки харчових мас у шлунку, що сприяє підвищенню шлункової секреції і розвитку виразок на слизовій оболонці;
2) розвитку антиперистальтики (зворотної перистальтики) шлунку, що обумовлює появу таких ознак диспепсії, як відрижка, нудота, блювота.
ЗО. ЗО. Що таке пілороспазм?
Шлороспазм — це спастичне скорочення пілоричної частини шлунку, одна з форм дискінезій шлунку гіпертонічного типу. Його спостерігають переважно у грудних дітей, частіше в перші тижні і місяці життя.
Вважають, що пілороспазм у дітей обумовлений функціональними розладами нервово-м'язового апарату пілоричної частини шлунку. Він буває головним чином у збудливих дітей, що зазнали внутрішньоутробної гіпоксії, що народилися в асфіксії, з ознаками пологової травми центральної нервової системи.
При пілороспазмі відзначають слабкий розвиток мускулатури кардіальної частини шлунку і більш виражений її розвиток у ділянці воротаря. Це сприяє легкому виникненню блювоти і зригувань.
30.31. Назвіть причини і значення гіпотонічних дискінезій шлунку.
Зменшення рухової (моторної) активності шлунку може бути обумовлено:
а) аліментарними факторами (жирна їжа);
б) зменшенням шлункової секреції (гіпоацидні гастрити);
в) зменшенням тонусу блукаючого нерва;
г) дією гастроінтестинальних гормонів, що пригнічують моторику шлунку, - гастро-інгібіторного пептиду, секретину та ін.;
ґ) видаленням пілоричної частини шлунку;
д) загальним ослабленням організму, виснаженням, гастроптозом (опущенням шлунку).
При гіпотонічних дискінезіях зменшується час перебування їжі в шлунку, що веде до порушення її перетравлювання. Дія неперетравлених компонентів їжі на рецептори слизової оболонки кишок викликає підвищення їхньої перистальтики і проноси.
30.32. Назвіть типи патологічної шлункової секреції.
Порушення шлункової секреції в експерименті виявляють шляхом визначення кількості шлункового соку і його складу після механічного, а потім хімічного впливу. Розрізняють такі типи патологічної шлункової секреції (рис. 143):
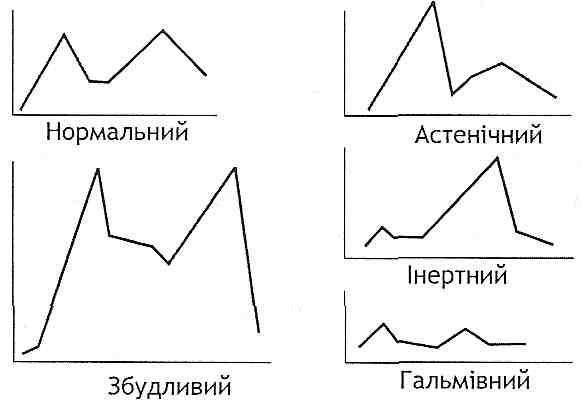
Рис. 143. Типи шлункової секреції
1) збудливий тип. Характеризується збільшенням секреції шлункового соку як на механічні, так і на хімічні стимули;
2) астенічний тип. Виявляється збільшенням секреції шлункового соку на механічні впливи і зменшенням - на хімічні;
3) інертний тип. Шлункова секреція зменшена при дії механічних стимулів і зростає при хімічній стимуляції;
4) гальмівний тип. Характеризується зменшенням секреції шлункового соку в умовах як механічної, так і хімічної стимуляції.
... ...
У людини залежно від кількості шлункового соку і його якісних характеристик J виділяють шлункову гіпер- і гіпосекрецію.
30.33. Чим виявляє себе шлункова гіперсекреція? Яке її значення?
Для шлунковоїгіперсекреціїхарактерно:
1) збільшення кількості шлункового соку як після приймання їжі, так і натще;
2) гіперацидітас і гіперхлоргідрія — відповідно збільшення загальної кислотності і вмісту вільної соляної кислоти в шлунковому соку;
3) збільшення перетравлювальної здатності шлункового соку.
Порушення травлення, пов'язані зі шлунковою гіперсекрецією, обумовлені тривалою затримкою вмісту у шлунку (воротар закритий, тому що нейтралізація дуже кислого хімусу, що надходить у дванадцятипалу кишку, вимагає багато часу). Ця обставина має такі наслідки:
1) у кишки надходить мало хімусу, що призводить до зменшення перистальтики кишок і розвитку закрепів;
2) у шлунку посилюються процеси бродіння і утворення газів. Це викликає появу відрижки й печії;
3) збільшується рухова активність шлунку — розвивається гіпертонус і гіперкінезія його гладкої мускулатури.
Утворення великої кількості активного шлункового соку є важливим чинником* що сприяє розвитку виразок у шлунку і дванадцятипалій кишці.
ЗО. 34. Як можна моделювати шлункову гіперсекрецію в експерименті?
Збільшення утворення і секреції шлункового соку моделюють за допомогою таких впливів.
I. Вплив на нервові механізми регуляції шлункової секреції:
а) збудження рецепторів слизової оболонки шлунку (введення в шлунок со-когінних екстрактів, алкоголю);
б) зменшення гальмівного впливу кори головного мозку на центри блукаючого нерва - метод "зіткнення" за І.Павловим (див. запит. 30.5);
в) електричне подразнення центрів блукаючого нерва або його еферентних волокон, що іннервують шлунок;
г) введення т-холіноміметиків.
II. Вплив на гуморальні механізми регуляції шлункової секреції:
а) введення гістаміну або фармакологічних агентів, що стимулюють його утворення в слизовій шлунку;
б) введення гастрину;
в) введення фармакологічних препаратів — стимуляторів Н2-рецепторів (ацетилсаліцилової кислоти, індометацину);
г) пригнічення утворення простагландинів, які в нормі гальмують секрецію соляної кислоти. З цією метою вводять глюкокортикоїди, ацетилсаліцилову кислоту.
Етіологія, патогенез виразкової хвороби шлунка та/або дванадцятипалої кишки. Етіологія, патогенез симптоматичних виразок шлунка та/або дванадцятипалої кишки.
30.35. Що таке виразкова хвороба? Яка її етіологія?
Виразкова хвороба — це хронічне рецидивуюче захворювання, що характеризується утворенням виразки в шлунку або дванадцятипалій кишці.
Етіологію виразкової хвороби донині остаточно не з'ясовано. Вважають, що у виникненні виразок шлунку і дванадцятипалої кишки мають значення такі фактори ризику.
1. Тривала психоемоційна перенапруга, як правило, негативного характеру (негативні емоції, конфліктні ситуації, почуття постійної тривоги, перевтома і т. п.), яка часто повторюється.
2. Стрес (див. розд. 33).
3. Спадкова схильність. Значення цього фактора підтверджує відносно висока (40-60 %) частота захворювання у батьків і родичів хворих, особливо молодого віку. Установлено, що у хворих з обтяженою спадковістю в слизовій оболонці шлунку у 1,5-2 рази більше парієтальних клітин (клітин, що продукують соляну кислоту), ніж у здорових. Ознаками генетичної схильності є також 0 (І) група крові, яку часто виявляють у хворих на виразку, дефіцит с^-антитрипсину і фукоглікопротеїдів.
4. Неправильне харчування - їжа всухом'ятку, нерегулярне приймання їжі, вживання грубої або гострої їжі, погане її пережовування, їда поспіхом, відсутність зубів, недостатній вміст у харчових продуктах білків і вітамінів.
5. Хронічний гастрит і дуоденіт з підвищеною секреторною активністю залоз слизової оболонки.
6. Мікробний фактор - Helicobacter (Campylobacter) pylori.
7. Шкідливі звички — куріння, уживання алкоголю.
30.36. Які існують теорії патогенезу виразкової хвороби?
1. Судинна теорія (Р. Вірхов). Відповідно до цієї теорії, виразки в шлунку і дванадцятипалій кишці виникають у результаті порушень кровопостачання їхніх стінок.
2. Механічна теорія (Л. Ашофф). Виникнення виразки пов'язано з травмуванням грубою їжею слизової оболонки шлунку в ділянці малої його кривизни.
3. Запальна теорія (Г. Конечний). В основі утворення виразок лежать запальні зміни слизової оболонки (гастрит та ерозії).
4. Пептична теорія (Е. Рігель). Пояснює виникнення виразки перетравлювальною дією шлункового соку на ділянки слизової, що найбільше зазнають впливу протеолітичних ферментів.
5. Нервово-вегетативна теорія (Г. Бергман). її прихильники вважають, що причиною утворення виразки є гіперсекреція шлункового соку, гіпермоторика шлунку і судинні порушення в ньому в осіб з конституціонально обумовленим переважанням тонусу парасимпатичної нервової системи.
6. Нервово-рефлекторна теорія (І. Греков, М. Стражеско). Пояснює виникнення виразки рефлекторними впливами на шлунок, наприклад, при хронічному апендициті, коліті, жовчнокам'яній хворобі.
7. Кортико-вісцеральна теорія (К. Биков, І. Курцин). Підкреслює провідну роль порушень умовнорефлекторної діяльності головного мозку (неврозів) в утворенні виразок.
8. Інфекційна теорія (Б. Маршали). Виразка у шлунку є результатом інфікування бактерією Helicobacter pylori (див. запит. 30.38.). Нині формується концепція, відповідно до якої утворення виразки в шлунку і дванадцятипалій кишці відбувається в результаті змін у співвідношенні факторів "агресії" і "захисту". До "факторів агресії" відносять підвищення кислотності і ферментативної активності шлункового соку в умовах порушення моторики шлунку і дванадцятипалої кишки, Helicobacter pylori. Зменшення захисних властивостей обумовлено зниженням продукції слизу, уповільненням процесів фізіологічної регенерації поверхневого епітелію, порушеннями місцевого кровообігу і нервової трофіки слизової оболонки.
30.37. Які виділяють патогенетичні варіанти виразки шлунку?
1. Екзогенна виразка.
2. Пептична виразка.
3. Трофічна виразка..
4. Гіпорегенераторна виразка.
30.38. Що таке екзогенна виразка шлунку? Що може бути її причиною?
Екзогенною називають виразку, що виникає в результаті безпосередньої дії на слизову оболонку шлунку ушкоджувальних факторів зовнішнього середовища.
В експерименті множинні виразки шлунку екзогенного походження моделюють за допомогою:
а) фізичних впливів - уведення в шлунок роздробленого скла (механічне ушкодження), окропу, рідкого азоту і т. п.;
б) хімічних впливів - уведення в шлунок сильних кислот і лугів.
У розвитку екзогенної виразки в людини найбільше значення мають мікробні фактори, зокрема Helicobacter pylori. Цей збудник потрапляє в шлунок через рот, занурюється в захисний шар слизу і прикріплюється до апікальної поверхні клітин епітелію. Найактивніше він заселяє вихідний відділ шлунку і цибулину дванадцятипалої кишки. Проникнення збудника в слизову викликає локальну імунну відповідь — антитіла, що утворилися, взаємодіють з поверхневими антигенами бактерій. Це у свою чергу спричиняє вихід у слизову поліморфноядерних лейкоцитів (нейтрофілів) - розвивається запалення (набряк, гіперемія), утворюються спочатку ерозії, які згодом переходять у виразку.
30.39. Що таке пептична виразка шлунку? Як її моделюють в експерименті?
Пептичною називають виразку, що виникає в результаті перетравлювальної дії шлункового соку на слизову оболонку шлунку і дванадцятипалої кишки.
Розрізняють два патогенетичних варіанти пептичної виразки: 1) виразка, що утворюється в результаті підвищення "агресивності" шлункового соку (збільшення кількості соку, його кислотності і перетравлювальної сили). В експерименті може виникати при моделюванні шлункової гіперсекреції (див. запит. 30.34);
2) виразка, обумовлена зменшенням захисних властивостей слизової оболонки. За цих умов шлунковий сік, що має нормальну "агресивність", чинить перетравлю-вальну дію на змінену слизову. Існує два підходи до моделювання в експерименті пептичної виразки цього виду:
а) порушення утворення слизу (введення глюкокортикоїдів, саліцилатів);
б) руйнування слизового бар'єра речовинами-детергентами (введення в шлунок жовчних кислот, відтворення дуодено-гастрального рефлюксу).
30.40. Що таке трофічні виразки шлунку? Як їх відтворюють в експерименті?
Трофічними називають виразки, які виникають у результаті порушення живлення слизової оболонки шлунку. До трофічних виразок відносять:
а) судинні виразки. Утворюються внаслідок порушень кровопостачання стінки шлунку. В експерименті їх моделюють перев'язуванням артерій, що живлять шлунок, введенням у шлункові артерії атофану — речовини, яка викликає склерозування стінки і облітерацію просвіту судин; введенням великих доз катехоламінів, що спричиняють спазм артерій та ішемію слизової шлунку;
б) нейрогенні виразки. Є результатом порушень нервової трофіки і розвитку нейро-дистрофічного процесу. В експерименті моделюють перетинанням нервів, що ін-нервують стінку шлунку.
30.41. Що таке гіпорегенераторні виразки шлунку? Чим вони можуть бути зумовлені?
Гіпорегенераторні виразки шлунку виникають у результаті порушення регенерації епітелію слизової оболонки. У нормі покривний епітелій і залозистий апарат слизової шлунку повністю оновлюється через кожні 5 діб. Якщо процеси регенерації з якихось причин порушуються, то спочатку утворюються поверхневі дефекти — ерозії, а потім і глибші — виразки.
Розвиток гіпорегенераторних виразок може бути зумовлений:
а) дією високих доз глюкокортикоїдів, що пригнічують біосинтез білка і клітинний поділ;
б) білковим голодуванням;
в) гіповітамінозами;
г) дією отрут — інгібіторів біосинтезу білків.
30.42. Чим виявляє себе шлункова гіпосекреція? Яке її значення?
Для шлунковоїгіпосекреціїхарактерні:
1) зменшення кількості шлункового соку натще і після приймання їжі;
2) зменшена або нульова кислотність шлункового соку (гіпо- або онацидітас), зменшення вмісту в ньому або повна відсутність вільної соляної кислоти (гіпо- або ахлоргідрія);
3) зменшення перетравлювальної здатності шлункового соку аж до ахілії (повного припинення утворення соляної кислоти і ферментів).
Зменшення шлункової секреції спричиняється до розладів травлення у всьому травному каналі. Це обумовлено тим, що в умовах недостатнього утворення шлунко- і вого соку воротар постійно відкритий і вміст шлунку швидко переходить у дванадця- І типалу кишку, де середовище внаслідок відсутності надходження зі шлунку соляної кислоти постійно лужне. Ця обставина гальмує утворення секретину, внаслідок чого зменшується секреція підшлункового соку й порушуються процеси порожнинного травлення в кишках. Недостатньо перетравлені компоненти їжі подразнюють рецептори слизової оболонки кишок, що призводить до посилення їхньої перистальтики, -розвиваються проноси.
Крім того, внаслідок відсутності соляної кислоти в шлунку посилено розмножується мікрофлора. З цим пов'язана активація процесів гниття й бродіння в шлунку і поява таких диспепсичних симптомів, як відрижка, обкладений язик та ін.
30.43. Як в експерименті можна моделювати шлункову гіпосекрецію?
І. Вплив на нервові механізми регуляції шлункової секреції:
а) блокада рецепторів слизової оболонки шлунку (застосування речовин, що викликають місцеву анестезію);
б) перетинання блукаючого нерва або його гілок (селективна ваготомія);
в) використання гангліоблокаторів (п-холінолітиків);
г) застосування ш-холінолітиків.
її. Вплив на гуморальні механізми регуляції утворення і секреції шлункового соку:
а) блокада Н -рецепторів (гістамінових), наприклад, циметидином;
б) застосування антагоністів гастрину - секретину, гастроінгібіторного пептиду та ін.
Порушення порожнинного травлення в кишках; причини, механізми, прояви. Розлади, пов'язані із секреторною недостатністю підшлункової залози. Панкреатити: види, причини; патогенез гострого панкреатиту. Панкреатичний шок.
30.54. Що таке синдром мальдигестії? Чим він виявляє себе?
Синдромом мальдигестії позначають порушення порожнинного травлення, обумовлені недостатнім надходженням в кишки травних ферментів, зокрема при панкреатичній гіпосекреції.
Цей синдром виявляє себе:
1) порушенням перетравлювання жирів (відсутність ліпази, фосфоліпаз). Не засво- | юється 60-80 % жиру, він виводиться з калом— з'являється симптом стеаторег\ (жир у калі);
2) розладами всмоктування жиророзчинних вітамінів — розвиваються ознаки гіповітамінозів А, Е, К;
3) порушенням перетравлювання білків (відсутність травних протеаз). Не засвою- | ється до 30—40 % харчового білка. У калі з'являється велика кількість м 'язових j волокон;
4) порушенням перетравлювання вуглеводів (відсутність амілази);
5) порушенням розщеплення нуклеїнових кислот (відсутність нуклеаз).
30.44. Які причини панкреатичної гіперсекреції? Чим вона може виявлятися?
Збільшення утворення і секреції підшлункового соку (панкреатична гіперсе-креція) може бути обумовлене:
1) підвищенням тонусу парасимпатичної нервової системи (блукаючого нерва);
2) збільшенням утворення і секреції гастроінтестинальних гормонів, що стимулюють секрецію води і гідрокарбонатів у складі підшлункового соку (секретин) і підвищують вміст у ньому травних ферментів (холецистокінін-панкреозимін).
Збільшення панкреатичної секреції поліпшує процеси порожнинного травлення, однак за деяких умов може сприяти розвитку гострого панкреатиту.
30.45. Що таке гострий панкреатит? Яка його етіологія?
Гострий панкреатит - це запалення підшлункової залози, що характеризується гострим перебігом.
Його виникнення і розвиток можуть бути обумовлені такими етіологічними факторами:
1) приймання великої кількості жирної їжі;
2) зловживання алкоголем і переїдання, що його супроводжує;
3) жовчні камені та поліпи протоки підшлункової залози;
4) механічне ушкодження підшлункової залози при травмах і хірургічних втручаннях;
5) інфекційні агенти (віруси епідемічного паротиту, коксакі, бактеріальна інфекція);
6) інтоксикації, у тому числі дія деяких лікарських засобів (імунодепресанти, тіази-ди та ін.).
30.46. Що є головною ланкою патогенезу гострого панкреатиту? Які розрізняють патогенетичні варіанти цього захворювання?
Провідним механізмом розвитку гострого панкреатиту будь-якої етіології є передчасна активація ферментів підшлункового соку в протоках підшлункової залози. Найбільше значення має активація трипсину, тобто утворення його із трипсиногену під дією активних протеолітичних ферментів клітинного (лізосомного) або кишкового походження. Особливістю активації ферментів у протоках підшлункової залози є те, що цей процес має лавиноподібний характер і, виникнувши в одному місці залози, швидко охоплює всі її протоки. Це обумовлено аутокаталітичними реакціями, під час яких молекули активних ензимів (трипсину, хімотрипсину) перетворюють неактивні ферменти (трипсиноген, хімотрипсиноген) в активні.
Таким чином, для ініціювання гострого панкреатиту потрібна "іскра", якою можуть стати навіть поодинокі молекули активних протеаз, що потрапили в протоки залози з ушкоджених клітин або кишок.
Залежно від джерела таких протеаз і механізмів їх потрапляння в протоки підшлункової залози розрізняють три патогенетичних варіанти гострого панкреатиту.
I. Первинно-альтеративний.
II. Гіпертензивний.
III. Рефлюксний.
ЗО. 4 7. Чим характеризується первинно-альтеративний варіант розвитку гострого панкреатиту?
Первинно-альтеративний варіант гострого панкреатиту розвивається в результаті ушкодження тканини підшлункової залози різними патогенними факторами. Це може бути механічне ушкодження (травма, операційні втручання), вплив хімічних агентів (отруєння лугами, кислотами) і біологічних чинників (віруси, бактерії); ішемічне або алергічне ушкодження.
Ініціаторами передчасної активації ферментів підшлункового соку у всіх цих випадках є лізосомні ферменти, що вивільняються з ушкоджених клітин залозистої тканини.
30.48. Що лежить в основі розвитку гіпертензивного варіанта гострого панкреатиту?
Гіпертензивний варіант гострого панкреатиту розвивається в результаті підвищення тиску панкреатичного соку в протоках підшлункової залози.
Залежно від причин виникнення гіпертензії розрізняють два різновиди цього варіанта:
1) гіперсекреторний панкреатит. Причиною підвищення тиску в протоках залози є різке збільшення секреції підшлункового соку (жирна їжа, алкоголь, що стимулю-
ють утворення секретину і холецистокінін-панкреозиміну), коли панкреатичні протоки не в змозі швидко пропустити весь сік, що утворився (відносна недостатність^ проток). У цьому випадку мають значення анатомо-фізіологічні особливості вивід-: них проток залози, тому панкреатит у такій формі може виникати не в усіх людей; 2) обтураційний панкреатит. Гіпертензія в протоках підшлункової залози виникає при нормальному рівні секреції, але при наявності перешкод (обтурації) відтоку соку (камені, здавлювання проток ззовні та ін.). Підвищений тиск соку є причиною ушкодження клітин, що вистилають протоки залози. Це призводить до виходу лізосомних ферментів і активації трипсиногену з наступною реалізацією аутокаталітичного механізму (див. запит. 30.46).
30.49. Які умови викликають рефлюксний варіант розвитку гострого панкреатиту?
Рефлюксний варіант гострого панкреатиту виникає в результаті закидання в протоки підшлункової залози вмісту дванадцятипалої кишки або жовчі.
Дуодено-шнкреатичний рефлюкс виникає при збільшенні внутрішньодуоде-нального тиску (кишкова непрохідність) або розслабленні сфінктера Одді. Ініціатором передчасної активації ферментів підшлункової залози в цьому випадку є енте-рокіназа - протеолітичний фермент, що утворюється клітинами слизової оболонки дванадцятипалої кишки.
Умовою виникнення жовчно-панкреатичного рефлюксу є наявність спільної кінцевої ділянки панкреатичної і загальної жовчної проток (спільна ампула). При цьому жовчні кислоти, що мають детергентну дію, при потраплянні в протоки підшлункової залози ушкоджують їхні клітини. З останніх виходять лізосомні ферменти, які і запускають процес активації панкреатичних ензимів. Крім того, жовчні кислоти можуть активувати ліпазу підшлункового соку, хоча є дані, що панкреатична ліпаза не потребує активації.
30.50. Опишіть патогенез місцевих змін у підшлунковій залозі при гострому панкреатиті.
Передчасна активація ферментів панкреатичного соку в протоках підшлункової залози викликає самоперетравлювання залозистої тканини. Так, під дією трипсину й хімотрипсину відбувається гідролітичне розщеплення тканинних білків і білкових компонентів клітин. Еластаза спричинює гідроліз еластину — складової частини базальних мембран судин. Фосфоліпаза, розщеплюючи фосфоліпіди клітинних мембран, обумовлює порушення їх бар'єрної функції, а ліпаза, гідролізуючи тригліцериди, викликає розвиток некрозу жирової тканини. Із фосфоліпідів мембран вивільняється арахідонова кислота, з якої утворюються простагландини. З крові в тканину залози надходить калікреїноген і а2-глобуліни. Під дією трипсину і хімотрипсину відбувається активація калікреїну і в кінцевому підсумку утворюються кініни.
Активні ферменти підшлункового соку, простагландини, кініни - усі ці фактори викликають вторинну альтерацію тканини підшлункової залози, підвищення проникності судин з розвитком набряку, геморагій; виникнення болю. Розвивається гостре запалення, що має ряд відмітних ознак. По-перше, воно є генеролізованим, тобто охо-
плює всю тканину залози; по-друге, надто сильною є альтерація (іноді розвивається повний некроз залози), по-третє, дуже характерні судинні зміни і пов'язана з ними ексудація (набряк, геморагії).
ЗО. 51. Які механізми лежать в основі розвитку панкреатичного шоку?
Панкреатичний шок є важким загальним проявом гострого панкреатиту і характеризується порушеннями загальної гемодинаміки (падінням артеріального тиску) та генералізованими розладами мікроциркуляції.
У патогенезі панкреатичного шоку мають значення два механізми.
I. Больовий механізм. Причиною різкого гострого оперізуючого болю, що виникає при панкреатиті, з одного боку, є набряк підшлункової залози (тиск на сонячне сплетіння), а з другого - дія активних травних ферментів (трипсин, фосфоліпаза та ін.) та біологічно активних речовин (кініни, простагландини) на нервові закінчення залози. Інтенсивний біль викликає спочатку збудження, а потім позамежне гальмування в життєво важливих центрах головного мозку. Це веде до пригнічення зовнішнього дихання і порушення системного кровообігу — розвивається больовий шок (див. розд. 12).
II. Гуморальний механізм. Він обумовлений ферментемією — надходженням у кров активних панкреатичних ферментів. Ферменти, що потрапили в кров, інак-тивуються природними інгібіторами протеаз (aj-інгібітор протеаз, антитромбін III, а2-макроглобулін, а2-антиплазмін, інтер-а-інгібітор трипсину та ін.). Однак якщо потужність інгібіторів недостатня і всі ферменти, що надійшли, не можуть бути інактивовані, то наступною подією є активація біохімічних систем крові. її зумовлює головним чином трипсин. При цьому активуються калікреїн-кінінова система, системи зсідання крові і фібринолізу. Кініни, що утворюються, викликають генералізоване розширення судин, яке веде до зменшення загального периферичного опору, з другого ж боку вони підвищують проникність судин, унаслідок чого рідка частина крові переходить у тканини (плазморагії) і, отже, зменшується об'єм циркулюючої крові. Зазначені зміни гемодинаміки обумовлюють падіння артеріального тиску. Активація системи зсідання і фібринолітичної системи є причиною розвитку ДВЗ-синдрому і пов'язаних з ним розладів мікроциркуляції (див. розд. 26.3).
30.52. Якими синдромами виявляє себе панкреатичний шок?
1. Гостра артеріальна гіпотензія (колапс). Обумовлена розширенням судин (зменшується загальний периферичний опір) і падінням об'єму циркулюючої крові (зменшується хвилинний об'єм серця).
2. ДВЗ-синдром. Пов'язаний з активацією системи зсідання крові і фібринолітичної системи, а також з підвищенням проникності кровоносних судин.
3.- Гіпоксичний синдром. Обумовлений:
а) циркуляторною гіпоксією — порушеннями загальної гемодинаміки (падіння артеріального тиску) і мікроциркуляції (ДВЗ-синдром);
б) дихальною гіпоксією—пригніченням діяльності дихального центру (позамежне гальмування життєво важливих центрів при сильному больовому синдромі);
в) гемічною гіпоксією, що виникає в результаті гемолізу еритроцитів, обумовленого панкреатичною фосфоліпазою. 4. Інтоксикаційний синдром. Пов'язаний з надходженням у кров продуктів аутолі-тичного розпаду тканини підшлункової залози.
30.53. Які причини викликають розвиток панкреатичної гіпосекреції? Яке її значення?
Причинами зменшення утворення і секреції підшлункового соку (панкреатичної гіпосекреції) можуть бути:
а) нейрогенне гальмування зовнішньосекреторної функції підшлункової залози, (зменшення тонусу блукаючого нерва, отруєння атропіном та ін.);
б) дуоденіт - запалення слизової дванадцятипалої кишки, унаслідок чого зменшується утворення стимуляторів панкреатичної секреції - секретину і холецистокі-1 нін-панкреозиміну;
в) порушення виведення підшлункового соку (закупорка проток, їх здавлювання);
г) зменшення кількості секреторних клітин (руйнування, хронічні панкреатити);
ґ) спадково обумовлена недостатність ентерокінази, внаслідок чого порушується початкова активація протеолітичних ферментів підшлункового соку (трипсину, хімотрипсину). Головним наслідком панкреатичної гіпосекреції є порушення порожнинного
травлення в кишках — розвиток синдрому мальдигестії.
Порушення всмоктування. Причини і механізми мальабсорбції, патогенез основних клінічних проявів.
30.61. Що таке синдром мальабсорбції? Чим можуть бути обумовлені порушення всмоктування?
Синдромом мальабсорбції називають симптомокомплекс, що виникає в результаті порушень усмоктування речовин у кишках.
Порушення всмоктування в кишках можуть бути обумовлені розладами, що виникають на трьох рівнях. Тому виділяють:
1) передентероцитарні порушення. Розвиваються внаслідок розладів процесів травлення, що передують усмоктуванню; ; 2) ентероцитарні. Виникають у результаті порушення діяльності епітеліальних клітин слизової кишок (ентероцитів); 3) постентероцитарні. Є наслідком порушення процесів, які забезпечують надходження речовин, що всмокталися, у внутрішнє середовище організму (кров, лімфу).
30.62. Які передентероцитарні порушення можуть лежати в основі розладів усмоктування в кишках?
1. Порушення рухової функції травного каналу.
2. Порушення порожнинного травлення (синдром мальдигестії). За походженням вони можуть бути гастрогенними, панкреатогенними, гепатогенними, ентероген-ними, дисрегуляторними, ятрогенними (пов'язаними з тривалим застосуванням антибіотиків та інших лікарських препаратів).
З. Порушення пристінкового травлення. Найчастіше зумовлені розладами утворення і вмонтовування ферментів у плазматичну мембрану мікроворсинок ентероцитів.
30.63. Що таке інтестинальні ферментопатії? Чим вони можуть виявлятися?
Інтестинальні ферментопатії — це спадково обумовлені порушення синтезу травних ферментів мікроворсинок, що забезпечують процеси пристінкового (мембранного) травлення.
Серед інтестинальних ферментопатій найчастіше бувають непереносність дисахаридів (лактози, сахарози, трегалози) і недостатність пептидаз (глютенова хвороба).
Непереносність дисахаридів обумовлена недостатністю дисахаридаз. У деяких регіонах Африки, Азії і Південної Америки 90 % населення не переносить молока і його не споживає. У представників зазначених етнічних груп нема ферменту лактази, що розщеплює лактозу молока. Через це нерозщеплена лактоза, накопичуючись на поверхні слизової, подразнює рецептори кишок і викликає профузні проноси.
Глютенову хворобу зумовлює недостатність пептидаз, вона характеризується не-переносністю продуктів, виготовлених зі злаків. Поширена в Європі, зокрема в Голландії. Глютен — білкова частина клейковини деяких злаків (пшениці, рису, ячменю, вівса) - складається з нешкідливого глютеїну і гліадину, який токсично діє на слизову тонкої кишки (викликає ентерит, проноси). При недостатності пептидаз гліадин не розщеплюється і чинить свою токсичну дію.
Кишкові дискінезії у дітей. Причини і механізми закрепів та проносу. Кишкова непрохідність: види, етіологія, патогенез. Особливості у дітей.
30.55. Що таке кишкові дискінєзії? Які існують їхні варіанти?
Кишковими дискінезіями називають порушення рухової (моторної) функції кишок.
Виділяють два варіанти кишкових дискінезій: гіперкінетичний і гіпокінетичний. \ Для першого характерно посилення перистальтики кишок, ритмічної сегментації і маятникоподібних рухів, що виявляється розвитком проносів (діареї).
Другий, навпаки, характеризується послабленням рухової активності кишок, унаслідок чого виникають закрепи.
30.56. Назвіть причини і значення гіперкінетичних дискінезій кишок.
Причинами кишкових дискінезій гіперкінетичного типу можуть бути:
а) підвищення збудливості рецепторів кишок до адекватних подразників під час розвитку запалення слизової оболонки кишок (ентерити, коліти);
б) дія на рецептори кишок незвичайних, патологічних подразників — неперетравленої їжі (наприклад, при ахілії), продуктів гниття й бродіння, токсичних речовин і т. п.;
в) збільшення збудливості центрів блукаючого нерва;
г) збільшення утворення деяких гастроінтестинальних гормонів, що посилюють перистальтику кишок, наприклад, мотиліну.
Наслідками кишкових дискінезій гіперкінетичного типу є:
а) розлади травлення (перетравлювання, всмоктування);
б) зневоднення;
в) видільний негазовий ацидоз (втрата гідрокарбонатів).
30.57. Чим виявляють себе гіпокінетичні дискінезії кишок? Які причини їх розвитку і значення?
Кишкові дискінезії гіпокінетичного типу виявляються зменшенням перистальтики кишок, що призводить до виникнення закрепів. Залежно від механізмів розвитку виділяють два види закрепів: спастичні й атонічні.
Спастичні закрепи виникають у результаті стійкого тривалого тонічного скорочення гладкої мускулатури кишок (спазму) і можуть бути обумовлені вісцеро-вісце-ральними рефлексами або дією токсичних факторів (наприклад, отруєння свинцем).
Причиною розвитку атонічних закрепів, пов'язаних зі зменшенням скорочувальної функції гладких м'язів кишок, можуть бути:
а) бідне харчування, низький вміст клітковини в харчових продуктах;
б) надмірне перетравлювання їжі в шлунку (наприклад, при шлунковій гіперсекреції);
в) вікові зміни рецепторного апарату кишок в осіб старечого віку, а також структурні зміни кишкової стінки при ожирінні;
г) зменшення тонусу блукаючого нерва;
ґ) порушення внутрішньокишкової іннервації, наприклад, при хворобі Гіршпрун-га- відсутність гангліонарних клітин ауербахового сплетіння в сигмоподібній і прямій кишках. Кишкові дискінезії гіпокінетичного типу призводять до:
1) розвитку кишкової аутоінтоксикації;
2) виникнення метеоризму;
3) утворення калових каменів;
4) у крайніх випадках може розвиватися кишкова непрохідність.
30.58. Що таке кишкова непрохідність? Як її класифікують?
Непрохідність кишок (ileus) - це захворювання, що характеризується порушенням проходження хімусу по кишках. Причиною цього можуть бути або обтурація, або здавлювання кишок, або порушення їхніх функцій.
Кишкову непрохідність класифікують у такий спосіб.
I. Механічна:
а) обтураційна. Розвивається внаслідок закупорки просвіту кишки пухлиною, каловими каменями, клубком гельмінтів;
в) странгуляційна. Є результатом здавлювання кишки ззовні (заворот, защемлення в грижових воротах, утворення вузлів). Особливістю цього виду непрохідності є здавлювання судин брижі, що порушує живлення стінки кишки аж до некрозу.
II. Динамічна:
а) спастична. Обумовлена спастичним скороченням гладких м'язів кишок;
б) паралітична. Розвивається внаслідок глибокого пригнічення рухової функції кишок.
ЗО. 59. Якими змінами в організмі виявляє себе непрохідність кишок? Який їх патогенез?
I. Больовий синдром. Розвивається внаслідок спастичного скорочення гладких м'язів, некрозу стінки кишки, розтягнення її рідиною. У результаті тривалого інтенсивного болю можуть з'являтися ознаки больового шоку (падіння артеріального тиску, розлади зовнішнього дихання), обумовлені позамежним гальмуванням судинорухового і дихального центрів після їх перезбудження.
II. Зневоднення. В умовах кишкової непрохідності відбувається накопичення великої кількості рідини в кишках вище місця перешкоди. Причиною цього є:
а) збільшення секреції травних залоз у відповідь на розтягування стінки кишки рідиною і газами;
б) перехід рідини з кровоносних судин у кишки (транссудація) внаслідок застою крові і збільшення проникності стінок судин;
в) порушення процесів всмоктування води.
Накопичення рідини в кишках призводить до збільшення внутрішньокишково-jo тиску, що обумовлює подразнення великої кількості рецепторів і виникнення блювоти. Що вище перешкода в кишках, то інтенсивніша блювота і більш виражене зневоднення. Ш. Порушення обороту травних ферментів. Збільшення внутрішньокишкового тиску призводить до закидання вмісту кишок (у тому числі і травних соків, ен-терокінази) у протоки підшлункової залози. Відбувається передчасна активація ферментів підшлункового соку, унаслідок чого розвивається гострий панкреатит з явищами панкреатичного шоку (див. запит. 30.49 і 30.54).
IV. Порушення кислотно-основного стану. Якщо при нестримній блювоті втрачаються в більшій мірі хлориди — розвивається негазовий алкалоз, якщо гідрокарбонати - негазовий ацидоз (див. розд. 25).
V. Кишкова аутоінтоксикація. Особливо виражена при низькій кишковій непрохідності. Виявляється ознаками недостатності печінки (див. розд. 31).
VI. Гострий перитоніт (запалення очеревини). Обумовлений мікробами, які проникають через некротизовану стінку кишки в порожнину очеревини. Перитоніт є чинником, що викликає і посилює біль та больовий шок, а також, зумовлю-
ючи рефлекторну блювоту, сприяє розвитку зневоднення, порушень кислотно-основного стану і розладів системної гемодинаміки.
VII. Розлади загального кровообігу і мікроциркуляції. Порушення системної гемодинаміки (падіння артеріального тиску, зменшення хвилинного об'єму серця, зменшення загального периферичного опору) є проявом больового і панкреатичного шоку, а також зневоднення. Згущення крові (гемоконцентрація) призводить до розладів мікроциркуляції.
VIII. Гіпоксія. Обумовлена розладами кровообігу і зовнішнього дихання.
IX. Порушення функції життєво важливих органів (нирок, серця, головного мозку). Зумовлені розвитком гіпоксії та інтоксикації.
30.60. Які причини викликають порушення дефекації? Чим можуть виявлятися такі розлади?
Причинами порушень випорожнення кишок (дефекації) можуть бути: . а) випадання впливу кори головного мозку на спинномозковий центр дефекації (переляк, страх);
б) ушкодження центру дефекації в попереково-крижовому відділі спинного мозку;
в) ураження периферичних нервів: ші. pelvici, hypogastrici;
г) розлади функції м'язів, що беруть участь у дефекації.
Порушення випорожнення кишок можуть виявлятися:
1) мимовільною дефекацією;
2) відсутністю позивів на дефекацію або, навпаки, частими помилковими позивами;
3) нетриманням калових мас.
Недостатність печінки: визначення поняття, принципи класифікації, причини виникнення, експериментальне моделювання.
31.2. Що таке недостатність печінки? Як її класифікують?
Недостатність печінки— цс патологічний стан, при якому діяльність цього органа не здатна забезпечити сталість внутрішнього середовища організму відповідно до його вимог.
Класифікують її в такий спосіб. І. Недостатність печінки може бути відносною і абсолютною.
Відносна виникає при первинному підвищенні навантаження на печінку, коли вимоги організму щодо підтримання гомеостазу перевищують функціональні можливості печінки.
Абсолютна недостатність розвивається при первинному ураженні печінки, внаслідок чого зменшуються її функціональні можливості і вона не здатна забезпечувати сталість внутрішнього середовища навіть у звичайних умовах. Відносна недостатність печінки згодом може переходити в абсолютну. Послідовність подій у цьому випадку така: підвищення навантаження на печінку → від-
носна недостатність печінки → порушення сталості внутрішнього середовища → вторинне ураження печінки → абсолютна печінкова недостатність.
II. Залежно від причин ушкодження гепатоцитів абсолютна недостатність печінки може бути:
а) печінково-клітинною',
б) холестатичною;
в) печінково-судинною.
III. Залежно від кількості функцій, які порушуються при ураженнях печінки, недостатність цього органа може бути тотальною (порушуються всі види функцій печінки) і парціальною (страждає одна або кілька функцій).
IV. За клінічним перебігом недостатність печінки може бути гострою і хронічною.
31.3. У яких випадках розвивається печінково-клітинна недостатність печінки? Які її причини? Як її моделюють в експерименті?
Печінково-клітинна недостатність розвивається внаслідок прямого ушкодження гепатоцитів патогенними агентами.
Причинами її виникнення можуть бути фізичні фактори (іонізуюча радіація), гепатотропні отрути (чотирихлористий вуглець, токсини блідої поганки, алкалоїди геліотропа), віруси інфекційного і сироваткового гепатиту. Останні викликають ураження печінки внаслідок прямої цитотоксичної дії (розмножуються в гепатоцитах) і утворення аутоантитіл на змінені вірусом власні білки печінкових клітин (аутоімун-ний механізм ушкодження).
В експерименті на тваринах печінково-клітинну недостатність моделюють повним або частковим видаленням печінки, а також за допомогою отрут, що їх вводять в організм (чотирихлористого вуглецю, хлороформу, тринітротолуолу та ін.).
31.4. Що є причиною розвитку холестатичноїнедостатності печінки? Як її відтворюють в експерименті?
Холестшпична недостатність розвивається внаслідок первинних розладів жовчоутворення і жовчовиділення. Найчастішою причиною її розвитку є механічна жовтяниця (див. запит. 31.25).
Ушкодження гепатоцитів в умовах тривалого холестазу (порушення виведення жовчі) обумовлено принаймні двома причинами:
а) механічною дією жовчі на печінкові клітини (здавлення, розрив мембран);
б) роз'єднанням окиснення і фосфорування (дія білірубіну і жовчних кислот на мі-тохондрії), у результаті чого виникає дефіцит АТФ, а потім і дегенеративні зміни в гепатоцитах.
В експерименті холестатичну недостатність моделюють перев'язуванням жовчовивідних проток.
31.5. У яких випадках розвивається печінково-судинна недостатність печінки? Як її можна моделювати в експерименті?
Печінково-судинна недостатність розвивається в результаті первинних порушень кровообігу в печінці. При цьому основним механізмом ушкодження гепатоцитів є гіпоксія.
Найчастішими причинами печінково-судинної недостатності є портальна гіпер- \ тензія та ішемія печінки.
В експерименті розлади кровообігу в печінці моделюють за допомогою таких методів:
а) накладання порто-кавальних анастомозів (фістула Екка, фістула Екка-Павлова);
б) перев'язування ворітної вени;
в) перев'язування печінкових вен;
г) накладання лігатури на печінкову артерію
Типові порушення вуглеводного, білкового, ліпідного, водно-електролітного обмінів, обміну вітамінів і гормонів, системні порушення в організмі при недостатності печінки.
31.7. Порушеннями яких функцій печінки може виявляти себе її недостатність ?
При недостатності печінки можуть порушуватися такі її функції.
I. Метаболічні — участь печінки у вуглеводному, жировому, білковому обміні, обміні вітамінів, гормонів і біологічно активних речовин.
II. Захисні — фагоцитарна і антитоксична функції печінки.
III. Екскреторні — утворення і виділення жовчі. Секреція жовчі обумовлює видільну і травну функції печінки.
IV. Гемодинамічні - участь печінки у підтриманні системного кровообігу.
31.8. Які порушення вуглеводного обміну можуть розвиватися при ураженнях печінки?
Участь печінки у вуглеводному обміні полягає головним чином у підтримуванні сталості концентрації глюкози в плазмі крові. Це досягається завдяки тому, що в печінці відбувається депонування глюкози у вигляді глікогену (на глікоген припадає близько 20 % маси печінки).
Можливі два принципово різні стани, при яких порушується підтримування печінкою сталості концентрації глюкози крові. І. Зменшення вмісту глікогену в печінці. До цього можуть спричинятися: а) ненадходження глюкози з кишок у печінку (голодування);
б) порушення перетворення харчових моносахаридів (фруктози, галактози) у глюкозу, що характерно для спадково обумовлених захворювань — фрукто-земії і галактоземії;
в) порушення глюконеогенезу (наприклад, при гіпофункції кори надниркових залоз);
г) порушення синтезу глікогену з глюкози, обумовлене зменшенням активності ферментів глікогенезу (наприклад, спадково обумовлене захворювання — аглікогеноз);
ґ) дефіцит АТФ, необхідного для транспорту глюкози в гепатоцити і реакцій біосинтезу глікогену. її. Порушення процесів вивільнення глюкози з глікогену і надходження її в кров. Такі порушення лежать в основі спадкових захворювань, що одержали назву глікогенозів.
Оскільки утворення глюкози з глікогену в гепатоцитах може відбуватися двома шляхами (фосфоролітичним і гідролітичним), то можливі дві групи глікогенозів.
1. Глікогенози, при яких порушується фосфоролітичне розщеплення глікогену. До них, зокрема, відносять дефіцит кінази фосфорилази, фосфорилази (хвороба Гер-ша), глюкозо-6-фосфатази (хвороба Гірке).
2. Глікогенози з розладами гідролітичного розщеплення глікогену в лізосомах гепа-тоцитів. У цю групу, зокрема, входять хвороба Помпе, хвороба Форбса-Корі, хвороба Андерсена.
Морфологічно глікогенози виявляються значним збільшенням вмісту глікогену в гепатоцитах. Однак цей глікоген не може бути джерелом глюкози крові.
Основним проявом порушень вуглеводної функції печінки є розвиток печінкової гіпоглікемії, що у важких випадках може призводити до гіпоглікемічної коми (див. розд. 20).
31.9. Які розлади жирового обміну можуть виникати при ураженнях печінки?
1. Порушення перетравлювання і всмоктування жирів у тонкій кишці при патології жовчоутворення і жовчовиділення (див. запит. 31.29);
2. Порушення синтезу тригліцеридів, фосфоліпідів, холестеролу, а також утворення ліпопротеїдів плазми крові (ЛПДНГ і ЛПВГ);
3. Надмірне утворення кетонових тіл (див. розд. 21);
4. Жирова дистрофія (жирова інфільтрація, жировий гепатоз) печінки.
31.10. Що таке жирова дистрофія печінки? Який її патогенез?
Жирова дистрофія печінки — це збільшення у кілька разів проти норми вмісту тригліцеридів у печінкових клітинах, унаслідок чого розвивається дифузне або осередкове ожиріння печінки.
Найчастішими причинами жирової дистрофії печінки є ожиріння, цукровий діабет, алкоголь, хронічні інфекції та інтоксикації, гепатотропні отрути, аліментарні фактори (білкове голодування, дефіцит у їжі так званих ліпотропних речовин — холіну, метіоніну).
У патогенезі жирової дистрофії печінки виділяють два механізми (рис. 144).
Рис. 144. Механізми розвитку жирової дистрофії печінки
I. Надмірне утворення тригліперидів з жирових кислот. Це може бути зумовлено:
а) збільшеним надходженням жирових кислот у печінку (гіперліпацидемія);
б) порушенням окиснення жирових кислот у гепатоцитах (гіпоксія, дефіцит ] коферментів, переважне використання для енергетичних потреб інших сполук, зокрема алкоголю).
II. Недостатнє виведення тригліцеридів з печінки в кров у складі ЛХІДНГ (ліпо- І протеїдів дуже низької густини). Причинами цього можуть бути:
а) порушення синтезу основних компонентів ліпопротеїдних часток (білкової частини — апопротеїну, фосфоліпідів);
б) порушення формування міцел ліпопротеїдів;
в) розлади процесу секреції ліпопротеїдів.
31.11. Які розлади білкового обміну можуть виникати при ураженнях печінки?
Ураження печінки можуть виявляти себе такими розладами білкового обміну:
1) порушеннями біосинтезу білків, у тому числі і білків плазми крові (розлади біло-ксинтетичної функції печінки);
2) порушеннями перетворення амінокислот (дезамінування, трансамінування, де-карбоксилювання);
3) порушенням утворення сечовини (див. розд. 22).
31.12. Що може бути причиною порушень білоксинтетичної функції печінки? Чим виявляють себе такі порушення?
Білоксинтетична функція печінки порушується при: 1) змінах структури генів, що несуть інформацію про будову відповідних білків (наприклад, афібриногенемія);
2) порушеннях процесів транскрипції, тобто синтезу інформаційної РНК на матриці ДНК (наприклад, дія токсинів блідої поганки, антибіотика актиноміцину D);
3) розладах процесів транскрипції, тобто зчитування інформації з інформаційної РНК у рибосомах (наприклад, дія антибіотика пуроміцину);
4) зменшенні кількості амінокислот, необхідних для біосинтезу білків (наприклад, білково-енергетична недостатність, дефіцит у їжі незамінних амінокислот);
5) дефіциті АТФ, енергія якого використовується в біосинтетичних процесах (гіпоксія, голодування, гіповітамінози В] В2, РР, дія інгібіторів ферментів, роз'єднання окиснення й фосфорування).
У печінці синтезується переважна більшість білків плазми крові: всі альбуміни, 75-90 % а-глобуліців, близько 50 % р-глобулінів. Тому найяскравіші прояви порушень білоксинтетичної функції печінки пов'язані з розладами утворення саме білків плазми крові. Такими проявами є гіпопротеїнемічний і геморагічний синдроми.
Гіпопротеїнемічний синдром виникає в результаті зменшення концентрації альбумінів у плазмі крові. Це призводить до зниження онкотичного тиску плазми, наслідком чого може бути розвиток набряків.
Геморагічний синдром (підвищена кровоточивість) є наслідком порушень синтезу білків - факторів зсідання крові (фібриногену, протромбіну, проконвертину, про-акцелерину).
31.13. Які порушення обміну вітамінів можуть розвиватися при ураженнях печінки?
При ураженнях печінки можуть розвиватися такі розлади обміну вітамінів:
1) порушення утворення біологічно активних форм вітамінів з вітамінів-попередни-ків (наприклад, тіамінпірофосфату з вітаміну В,);
2) порушення депонування вітамінів у печінці (наприклад, вітаміну В12);
3) розлади всмоктування жиророзчинних вітамінів у результаті порушень функції утворення жовчі в печінці.
Клінічно всі зазначені розлади виявляють себе розвитком ознак відповідних гіповітамінозів.
31.14. Як порушується обмін мікроелементів при ураженнях печінки?
При захворюваннях печінки порушуються: а) депонування заліза (у формі феритину), міді, цинку, кобальту, молібдену, марганцю та ін.;
6) синтез транспортних білків, що забезпечують перенесення мікроелементів в організмі (трансферину, церулоплазміну);
в) екскреція мікроелементів з жовчю.
31.15. Які порушення обміну гормонів і біологічно активних речовин можуть супроводжувати розвиток недостатності печінки?
У печінці відбувається інактивація багатьох гормонів і біологічно активних речовин.
Інактивації зазнають усі стероїдні гормони (глюкокортикоїди, мінералокортико-їди, жіночі й чоловічі статеві гормони), тиреоїдні гормони, інсулін. При порушенні функції гепатоцитів зменшується інтенсивність перетворення зазначених гормонів, тому їхня концентрація в крові зростає і з'являються ознаки гіперфункції відповідних ендокринних залоз (вторинного гіперальдостеронізму, гіпертиреозу та ін.; див. розд. 33). Подібні ознаки можуть бути пов'язані і з порушенням синтезу в печінці транспортних білків (наприклад, транскортину), що зв'язують вільні гормони.
Печінка є органом, де відбувається руйнування багатьох біологічно активних речовин, і зокрема — біогенних амінів: катехоламінів, гістаміну, серотоніну. У зв'язку з цим у разі порушення функції гепатоцитів концентрація зазначених біологічно активних речовин у крові може зростати. У результаті розвиваються ознаки активації симпатоадреналової системи (підвищення артеріального тиску, тахікардія та ін.), підвищеного утворення гістаміну (свербіж шкіри, розвиток виразок у травному каналі).
Причини, механізми, клінічні прояви недостатності антитоксичної функції печінки. Теорії патогенезу печінкової коми.
31.16. Чим можуть виявляти себе порушення захисної (бар'єрної) функції печінки?
Захисна функція печінки складається з фагоцитарної й антитоксичної функцій.
Фагоцитарну функцію здійснюють особливі зірчасті ендотеліальні клітини (клітини Купфера), що належать до системи мононуклеарних фагоцитів. Можливі розлади фагоцитарної функції цих клітин описано в розд. 8.
Антитоксична функція властива гепатоцитам. Вона полягає в інактивації кінцевих продуктів обміну речовин (сечовиноутворення) і екзогенних речовин, що надходять з кишок (фенол, крезол, індол, скатол, аміни), а також з навколишнього середовища. Детоксикація різних речовин здійснюється за допомогою різних хімічних реакцій (синтезу, окиснення, відновлення, гідролізу, метилювання, кон'югації), що відбуваються в ендоплазматичному ретикулумі, мітохондріях, пероксисомах і цитоплазмі печінкових клітин.
Порушення антитоксичної функції печінки виявляються ознаками інтоксикації, що уражає насамперед центральну нервову систему. Тому комплекс змін, що виникають, одержав назву синдрому гепатоцеребральної недостатності.
31.17. Які фактори можуть викликати розвиток розладів антитоксичної функції печінки?
До порушень антитоксичної функції печінки можуть спричинятися:
1) зменшення кількості функціонуючих гепатоцитів (наприклад, при масивних не- ' крозах печінки). Так, установлено, що синтез сечовини з аміаку порушується при ураженні 80 % і більше паренхіми печінки;
2) зменшення активності ферментів, що беруть участь у реакціях детоксикації. Воно може бути зумовлено генетичними дефектами відповідних ферментів (наприклад, спадкове порушення ферментів синтезу сечовини) або набутими порушеннями їх утворення (розлади білоксинтетичної функції печінки);
3) зменшення концентрації речовин, що беруть участь в інактивації токсичних продуктів (кисню, глюкуронової і сірчаної кислот, гліцину, таурину, цистеїну та ін.);
4) дефіцит АТФ. При цьому можуть порушуватися процеси надходження й виведення з гепатоцитів токсичних речовин, а також реакції синтезу й кон'югації, які вимагають витрат енергії
31.21. Який патогенез печінкової коми?
Центральне місце в патогенезі печінкової коми належить надходженню в кров церебротоксичних речовин. Про це, зокрема, свідчить той факт, що у хворих у стані | печінкової коми в багато разів збільшується вміст зазначених сполук у крові. Крім J того, в експерименті показано, що введення тваринам церебротоксичних речовин су- ] проводжується розвитком ознак печінкової коми.
Механізми дії церебротоксичних сполук на центральну нервову систему досить І складні. Велике значення в патогенезі печінкової коми можуть мати такі зміни.
1. Порушення синаптіїчної передачі. Цілий ряд церебротоксичних речовин (низь-комолекулярні жирові кислоти, тирамін та ін.) є несправжніми нейромедіатора-ми. Вони, накопичуючись в тканинах головного мозку, або заміщають нормальні медіатори нервової системи, або порушують їх утворення з попередників. Такі зміни призводять до порушень передавання нервових імпульсів і, як наслідок, | до порушень міжнейронних взаємодій та розладів інтегративних функцій цен- ] тральної нервової системи. Крім того, у тканинах головного мозку зростає вміст ГАМК - гальмівного медіатора центральної нервової системи. Збільшення концентрації цієї речовини обумовлене її утворенням у кишках під дією мікрофлори, а також порушеннями процесів інактивації в печінці.
2. Порушення енергетичного обміну, що призводять до дефіциту АТФ. Провідна роль у розвитку таких порушень належить аміаку (рис. 145). Накопичуючись у великих кількостях, аміак зв'язується з глютаміновою і а-кетоглютаровою кислотами, перетворюючи їх в кінцевому підсумку в глютамін. Оскільки а-кетоглю-тарова кислота є одним із центральних метаболітів циклу Кребса, зв'язування її з аміаком веде до порушення функціонування цього метаболічного шляху і, як наслідок, до порушення реакцій ресинтезу АТФ.
Зменшення вмісту АТФ у нервових клітинах призводить до розладів процесів активного транспорту катіонів, порушення генерації нервових імпульсів, змін величини мембранного потенціалу.
3. Порушення функції клітинних мембран у результаті прямої дії церебротоксичних речовин. Такий їхній вплив виявляється насамперед розладами роботи Na-К-насосів, унаслідок чого падає мембранний потенціал і стають неможливими ] генерація і проведення нервових імпульсів.
Рис. 145. Церебротоксична дія аміаку
4. Розвиток метаболічного ацидозу і пов'язаних з ним порушень обміну електролітів. Ацидоз при печінковій комі обумовлений церебротоксичними речовинами — кислотами (піровиноградною і молочною кислотою, амінокислотами та їхніми похідними, низькомолекулярними жировими кислотами).
Недостатність екскреторної функції печінки: причини, механізми, клінічні прояви. Порушення обміну жовчних пігментів при різних видах жовтяниць. Холемічний і ахолічний синдроми.
31.22. Якими синдромами можуть виявляти себе порушення екскреторної функції печінки ?
Сутність екскреторної функції печінки полягає в утворенні і виділенні жовчі. Завдяки цьому здійснюється:
1) виведення з організму продуктів метаболізму (білірубіну) і надлишку деяких речовин (холестеролу);
2) участь печінки в процесах травлення (емульгування, перетравлювання і всмоктування жирів).
Порушення екскреторної функції печінки можуть виявляти себе такими синдромами: жовтяниця, холемічний синдром, ахолічний синдром.
31.23. Як у нормі відбувається обмін жовчних пігментів?.
Основний жовчний пігмент - білірубін — являє собою кінцевий продукт обміну гема. Головним джерелом білірубіну крові є гемоглобін еритроцитів.
Утворення білірубіну з гема відбувається в клітинах системи мононуклеарних фагоцитів (макрофагах селезінки, червоного кісткового мозку, клітинах Купфера в печінці), де фагоцитовані еритроцити зазнають гемолізу. Білірубін, що утворився,
нерозчинний у воді, тому його транспорт у печінку здійснюється у зв'язаному з білками вигляді. Такий білірубін одержав назву непрямого, оскільки дає реакцію з діа-зореактивом Ерліха тільки після попереднього осадження білків. Непрямий білірубін з крові надходить у печінку, де відбуваються три процеси, важливі з погляду обміну жовчних пігментів:
1) захоплення гепатоцитами зв 'язаного з білками (непрямого) білірубіну. Його забезпечують специфічні білкові рецептори плазматичної мембрани печінкових клітин;
2) кон'югація білірубіну з глюкуроновою кислотою, внаслідок чого утворюються глюкуроніди білірубіну (прямий білірубін);
3) екскреція прямого білірубіну у складі жовчі.
Усі зазначені процеси (захоплення, кон'югація і екскреція) вимагають витраті енергії АТФ.
Екскретований з жовчю білірубін у жовчному міхурі і тонкій кишці під дією ферментів мікрофлори перетворюється в уробіліноген. Основна кількість уробіліноге-ну в товстій кишці далі перетворюється в стеркобіліноген, що виводиться з калом і частково з сечею, всмоктуючись у кров у ділянці нижнього і середнього гемороїдальних сплетінь прямої кишки.
Друга, набагато менша частина уробіліногену бере участь у так званому печінково-кишковому кругообігу — всмоктується в тонкій кишці, потрапляє в печінку, частково зазнає окиснення, а частково знову надходить у жовчовивідні шляхи і у кишки.
31.24. Що таке жовтяниця? Які існують її види?
Жовтяниця (icterus) — це синдром, обумовлений збільшенням рівня білірубіну в крові, що виявляється жовтим забарвленням шкіри і слизових оболонок. Виділяють три види жовтяниці.
1. Гемолітична (надпечінкова) жовтяниця. Виникає в результаті гемолізу еритроцитів і збільшеного утворення білірубіну в клітинах системи мононуклеарних фагоцитів.
2. Паренхіматозна (печінкова) жовтяниця. її розвиток пов'язаний з ураженням печінки.
3. Механічна (рбтураційна, або підпечінкова) жовтяниця. Виникає в результаті порушення відтоку жовчі по жовчовивідних шляхах.
31.25. Який механізм розвитку гемолітичної жовтяниці? Які зміни пігментного обміну характерні для цього виду жовтяниці?
Гемолітична жовтяниця розвивається внаслідок гемолізу еритроцитів (див. розд. 26.1). Посилений фагоцитоз еритроцитів або самого гемоглобіну, що вивільнився зі зруйнованих еритроцитів, спричиняється до утворення у фагоцитах великої кількості білірубіну, який, зв'язуючись з білками, надходить у кров, а потім і в печінку. Гепатоцити при цьому зазнають підвищеного навантаження, перетворюючи великі кількості непрямого білірубіну в прямий і екскретуючи останній у складі жовчі. У цьому пояснення високого вмісту стеркобіліногену в калі (гіперхолічний кал) і в сечі. У зв'язку з тим, що непрямий білірубін не фільтрується в нирках (бо зв'язаний з білками), його нема в сечі.
З урахуванням цього основними ознаками порушення пігментного обміну при гемолітичній жовтяниці є:
а) збільшення вмісту непрямого білірубіну в крові;
б) збільшення вмісту стеркобіліногену в калі (гіперхолічний кал);
в) збільшення вмісту стеркобіліногену в сечі;
г) поява в сечі уробіліногену (у зв'язку з тим, що печінка не в змозі окислити великі кількості цієї речовини, яка надходить із кишок).
31.26. Які існують різновиди паренхіматозної (печінкової) жовтяниці? Як змінюється обмін жовчних пігментів при кожному з них?
В основі розвитку паренхіматозної (печінкової) жовтяниці лежать ізольовані або комбіновані порушення захоплення, кон'югації і екскреції білірубіну клітинами печінки.
Виділяють такі різновиди паренхіматозної жовтяниці:
1. Печінково-клітинна жовтяниця. Характеризується порушеннями всіх трьох процесів, що відбуваються в гепатоцитах: захоплення, кон'югації і екскреції білірубіну. Виникає при ушкодженні гепатоцитів (наприклад, вірусний гепатит), при дефіциті АТФ. При цьому внаслідок загибелі печінкових клітин утворюються сполучення між жовчними і кровоносними капілярами. У результаті жовч потрапляє в кров (холемія), а разом з нею і прямий білірубін. У клітинах, які не загинули, але ушкоджені, порушується захоплювання непрямого білірубіну й екскреція жовчі. Вона починає виділятися не тільки в жовчні капіляри, але й у кров. При цьому зменшується надходження жовчі в кишки (гіпохолія).
У зв'язку із зазначеними порушеннями виникають такі зміни показників пігментного обміну:
а) збільшення вмісту в крові непрямого білірубіну (порушується його захоплювання гепатоцитами);
б) збільшення вмісту в крові прямого білірубіну (результат надходження жовчі У кров);
в) зменшення вмісту стеркобіліногену в калі (гіпохолічний кал);
г) поява в сечі білірубіну;
ґ) зменшення або повна відсутність стеркобіліногену в сечі. Крім того, у крові і сечі виявляють жовчні кислоти (холалемія і холалурія).
2. Печінкові жовтяниці з ізольованими порушеннями процесів, що забезпечують виведення білірубіну з організму. Вони можуть бути обумовлені: ^
а) порушеннями захоплення непрямого білірубіну (синдром Жільбера — спадково обумовлений дефіцит рецепторів до білок-білірубінового комплексу). Виявляється збільшенням вмісту непрямого білірубіну в крові і відсутністю стеркобіліногену в калі і сечі;
б) розладами кон 'югації білірубіну (фізіологічна жовтяниця новонароджених, синдром Кріглера—Найяра). Вони найчастіше пов'язані з набутим або спадковим дефіцитом ферменту глюкуронілтрансферази. Виявляються збільшенням вмісту непрямого білірубіну в крові і зменшенням вмісту стеркобіліногену в калі і сечі;
в) порушеннями екскреції білірубіну (синдром Дабіна-Джонсона, синдроїЛ Ротора). їх причиною є дефекти (найчастіше спадкові) систем транспорту білірубіну і жовчі з гепатоцитів у жовчні капіляри. Виявляють себе збільшенням вмісту прямого білірубіну в крові, появою в крові й сечі жовчних кислот, білірубінурією, зменшенням вмісту або повною відсутністю стерко-біліногену в калі і сечі.
31.27. Які причини і патогенез механічної жовтяниці? Дайте характеристику порушень пігментного обміну при цьому виді
жовтяниці.
Механічна (підпечінкова) жовтяниця розвивається в результаті механічної пе- І решкодй^відтоку жовчі. Це може бути:
1) здавлення жовчовивідних шляхів ззовні (пухлина головки підшлункової залози, дія рубця);
2) їх закупорка каменем, гельмінтами, густою жовчю.
Механічне перешкоджання відтоку жовчі призводить до застою й підвищення тиску жовчі, розширення й розриву жовчних капілярів і надходження жовчі в кров як прямо, так і через лімфатичні шляхи.
У зв'язку з цим виникають такі зміни показників обміну жовчних пігментів:
а) збільшується вміст у крові прямого білірубіну (гіпербілірубінемія);
б) у крові з'являються жовчні кислоти (холалемія);
в) збільшується вміст у крові холестеролу (гіперхолестеролемія). З'являються моди- j фіковані ліпопротеїди (ліпопротеїд X), що мають атерогенні властивості;
г) у сечі з'являється білірубін (білірубінурія), унаслідок чого вона набуває темного забарвлення ("колір пива"), крім того в ній виявляють жовчні кислоти (холалурія). Із сечі зникає стеркобіліноген;
ґ) у калі нема стеркобіліногену (безбарвний кал).
Зазначені зміни пігментного обміну зумовлюють розвиток двох важливих клінічних синдромів, характерних для механічної жовтяниці: холемічного і ахолічного.
31.28. Що таке холємічний синдром? У яких випадках він виникає? Чим виявляє себе?
Холємічний синдром (синдром холестазу) обумовлений надходженням компонентів жовчі (жовчних кислот, прямого білірубіну, холестеролу) у кров у зв'язку з порушенням формування і відтоку жовчі.
Він закономірно виникає при механічній жовтяниці, а також деяких формах печінкової жовтяниці (печінково-клітинній, печінковій жовтяниці, обумовленій порушеннями екскреції жовчі).
Походження основних проявів синдрому: 1. Поява в крові жовчних кислот - холалемія. Цим зумовлені такі порушення:
а) розлади діяльності центральної нервової системи, що виникають як наслідок загальнотоксичної дії жовчних кислот (загальна астенія, дратівливість, що змінюється депресією; сонливість удень і безсоння вночі; головні болі, стомлюваність);
б) артеріальна гіпотензія, брадикардія. їхній розвиток обумовлений підвищенням тонусу блукаючого нерва і прямою дією жовчних кислот на синусно-передсердний вузол та кровоносні судини;
в) свербіж шкіри, що виникає в результаті подразнення нервових закінчень жовчними кислотами;
г) множинні ушкодження і загибель клітин, обумовлені детергентною дією жовчних кислот. Цим, зокрема, пояснюють гемоліз еритроцитів, запалення і некрози в різних органах і тканинах (печінковий некроз, перитоніт, гострий панкреатит та ін.);
ґ) поява жовчних кислот у сечі (холалурія).
2. Надходження у кров білірубіну. Ця обставина викликає появу жовтого забарвлення шкіри і слизових оболонок, тобто власне жовтяницю.
3. Збільшення вмісту в крові холестеролу. Це з невідомих поки що причин зумовлює появу аномального ліпопротеїду X, що має атерогенну дію.
31.29. Що таке ахолічний синдром? Чим він виявляється?
Ахолічним називають синдром, обумовлений ненадходженням жовчі в кишки у зв'язку з порушеннями її формування і відтоку. Для цього синдрому характерні:
1. Розлади перетравлювання і всмоктування жирів. Обумовлені порушенням процесів емульгування жирів, зменшенням активності панкреатичної ліпази, що активується жовчю; порушенням утворення міцел, що всмоктуються в тонкій кишці. Наслідком зазначених змін є:
а) поява жиру в калі — стеаторея;
б) розлади всмоктування жиророзчинних вітамінів, у результаті чого розвиваються гіповітамін
ози А, Е, К;
в) зменшення надходження в організм ненасичених жирових кислот, необхідних для побудови фосфоліпІдів клітинних мембран.
2. Порушення рухової функції кишок — ослаблення перистальтики і зменшення тонусу кишок (закрепи).
3. Посилення процесів гниття і реакцій бродіння в кишках у результаті зменшення бактерицидної дії жовчі. Це призводить до збільшення навантаження на антитоксичні системи печінки.
4. Зміни з боку калу - знебарвлення, стеаторея.
31.30. Що таке дисхолія? Які її причини? Який механізм виникнення жовчних каменів ?
Дисхолія — це порушення фізико-хімічних властивостей жовчі, внаслідок чого вона набуває літогенних властивостей, тобто здатності утворювати камені (конкременти) у жовчному міхурі й жовчних протоках. Результатом цього є розвиток жов-чноком 'яної хвороби.
Дисхолія і жовчні камені виникають унаслідок взаємодії багатьох чинників, серед яких: а) спадкова схильність;
б) нераціональне харчування;
в) порушення обміну речовин;
г) інфекційно-запальні процеси в жовчному міхурі і жовчних протоках; ґ) застій жовчі (холестаз).
Одним з основних механізмів виникнення літогенної жовчі є зниження холато-холестеролового і лецитин-холестеролового індексів (відношення жовчних кислот і лецитину до холестеролу жовчі). Це може бути зумовлено зменшенням печінково-кишкового кругообігу жовчних кислот при патології кишок і зміні їхньої мікрофлори, пригніченням синтезу жовчних кислот у печінці, прискоренням їх всмоктування слизовою оболонкою запаленого жовчного міхура, зменшенням вмісту лецитину і збільшенням синтезу холестеролу. При зменшенні концентрації жовчних кислот і лецитину, що забезпечують завислий стан холестеролу, останній випадає в осад і започатковує утворення холестеролових каменів.
Інфекція, застій жовчі також сприяють процесу утворення каменів, тому що супроводжуються зміною властивостей жовчі — зміщенням рН у кислий бік, зниженням розчинності солей, випаданням їх в осад, коагуляцією білків із клітин, що розпадаються. Крім холестеролових, утворюються пігментні (при гемолізі еритроцитів), вапняні і складні камені (наприклад, холестеролово-пігментно-вапняні). Камені обумовлюють порушення жовчовиділення і розвиток механічної жовтяниці.
31.31. Які причини, механізми розвитку і значення дискінезій жовчного міхура і жовчних проток?
Існує три варіанти порушень скоротливої функції жовчного міхура і жовчних проток (дискінезій).
1. Гіпертонічний (гіперкінетичний). Виявляється підвищенням тонусу гладких м'язів жовчного міхура і міхурової протоки, а також спазмом сфінктера Одді. Причиною його розвитку може бути підвищення тонусу блукаючого нерва або збільшення секреції холецистокінін-панкреозиміну.
2. Гіпотонічний (гіпокінетичний). Характеризується зменшенням тонусу жовчного міхура й міхурової протоки, сфінктер Одді постійно розслаблений. Виникає при зменшенні тонусу блукаючого нерва або при пригніченні утворення холецистокінін-панкреозиміну.
3. Змішаний варіант. Виявляє себе різноспрямованими змінами тонусу жовчного міхура й міхурової протоки, з одного боку, і сфінктера Одді —з другого. При цьому можливі два типи змін:
а) спазм сфінктера Одді і зменшення тонусу жовчного міхура і міхурової протоки;
б) розслаблення сфінктера Одді і підвищення тонусу жовчного міхура і міхурової протоки.
Порушення скоротливої функції жовчного міхура і жовчних проток спричиняється до розвитку больового синдрому, розладів жовчовиділення, а отже, і травлення.
Порушення гемодинамічної функції печінки. Синдром портальної гіпертензії: етіологія, патогенез, клінічні прояви.
31.32. Які функції печінки відносять до гемодинамічних? Чим виявляють себе розлади цих функцій?
Гемодинамічними називають функції печінки, які забезпечують її участь у здійсненні системного кровообігу. До них відносять:
а) колекторну функцію. Печінка збирає через систему ворітної вени кров з великого басейну- від органів черевної порожнини. Через печінку проходить 30-35 % хвилинного об'єму крові, що становить 1,5—1,8 л/хв;
б) депонування крові. У печінці може міститися до 700 мл крові, тимчасово виведеної з кровообігу. При необхідності (наприклад, після крововтрати) ця кров може бути мобілізована;
в) участь у підтриманні тонусу кровоносних судин через синтез білків, що є попередниками біологічно активних речовин — регуляторів артеріального тиску. Ідеться, зокрема, про синтез ангіотензиногену, з якого утворюється ангіотензин II.
Порушення гемодинамічних функцій печінки виявляють себе розвитком синдрому портальної гіпертензії.
31.33. Що таке синдром портальної гіпертензії? Які його причини? Чим він виявляється?
Синдром портальної гіпертензії розвивається в результаті порушення відтоку крові з органів черевної порожнини по судинах системи ворітної вени.
Залежно від того, де знаходиться перешкода відтоку крові, виділяють такі форми портальної гіпертензії:
1) підпечінкову — перешкода в стовбурі або великих гілках ворітної вени (емболи, здавлення пухлиною);
2) внутрішньопечінкову — перешкода в самій печінці (тривалий спазм гладком'язо-вих сфінктерів синусоїдів; здавлення дрібних печінкових вен вузлами регенеруючих гепатоцитів при ушкодженні печінки, її цирозі);
3) надпечінкову — перешкода локалізована у позаорганних відділах печінкових вен або в нижній порожнистій вені проксимальніше місця впадіння в неї печінкових вен. Сюди ж відносять портальну гіпертензію, що виникає при збільшенні тиску в системі нижньої порожнистої вени в умовах недостатності правого шлуночка серця.
Основні прояви синдрому портальної гіпертензії. 1. Здійснення колатерального кровообігу як результат розкриття портокавальних анастомозів. Це зумовлює розвиток таких ознак:
а) варикозне розширення вен стравоходу і кардіальної частини шлунку;
б) шлунково-кишкові кровотечі, причина яких ушкодження варикозно розширених вен;
в) розширення підшкірних вен передньої грудної і черевної стінки ("голова медузи");
г) скидання крові з ворітної вени в порожнисті в обхід печінки, що викликає інтоксикацію, а у важких випадках — розвиток екзогенної (портокавальної, або шунтової) печінкової коми.
2. Гепато-лієиальний синдром. Його важливими складовими є спленомегалія і пі перспленізм.
Спленомегалія — це збільшення розмірів селезінки. Вона виникає в результаті за-] стою крові.
Гіперспленізм — збільшення функціональної активності селезінки- виявляється посиленим руйнуванням формених елементів крові. Характеризується анемією, лейкопенією і тромбоцитопенією. В основі цього явища лежить збільшення фаго-І цитарної активності макрофагів селезінки в умовах уповільнення циркуляції кровя
3. Асцит (див. запит. 31.34).
4. Гепато-ренальний синдром. Виявляє себе порушеннями фільтраційної здатності ниркових клубочків при збереженні функцій канальцевого епітелію. Причиною цього, цілком імовірно, є зменшення тканинного кровообігу у зв'язку зі зменшен-І ням об'єму циркулюючої крові і зміною тонусу кровоносних судин, що спостері-1 гається при розладах гемодинамічних функцій печінки.
З1.34. Які механізми розвитку асциту?
Асцитом називають значне скупчення вільної рідини (як правило, транссудату) у черевній порожнині.
Причинами асциту можуть бути:
а) портальна гіпертензія різного походження;
б) набряки при хронічній недостатності серця, захворюваннях нирок, аліментарнім дистрофії;
в) порушення відтоку лімфи грудною протокою (її поранення, здавлення);
г) ураження очеревини пухлинним або туберкульозним процесом (асцит-перитоніт).
Асцитична рідина за своїм характером буває звичайно серозною, значно рідше -геморагічною.
У патогенезі асциту мають значення такі механізми:
1) гідростатичний. Пов'язаний з підвищенням тиску крові в капілярах судин ворітної системи;
2) онкотичний. Обумовлений зменшенням білоксинтетичної функції печінки, внаслідок чого розвивається гіпопротеїнемія й падає онкотичний тиск крові;
3) затримка натрію в організмі. Пов'язана зі збільшенням вмісту альдостерону в крові. Це у свою чергу обумовлено активацією ренін-ангіотензинної системи (застій крові в судинах ворітної системи → зменшення венозного повернення -* падіння хвилинного об'єму серця → гіпоксія нирок → вивільнення реніну). Крімj того, у зв'язку з розладами метаболічних функцій печінки може порушуватися! інактивація альдостерону, що рівнозначно його гіперпродукції;
4) лімфогенниймеханізм. У зв'язку з порушенням лімфовідтоку відбувається перехід багатої білками лімфи в черевну порожнину. Це викликає підвищення онкотичного тиску рідини черевної порожнини з наступним виходом у неї води із кровонос-і них судин та інтерстиціального простору
Причини та механізми порушень процесів фільтрації, реабсорбції та секреції в нирках. Функціональні проби для з'ясування порушень ниркових функцій.
У нирках відбувається дві групи процесів, що забезпечують підтримання гомеостазу, - сечоутворення (екскреторна функція) і вивільнення в кров гормонів, ферментів, біологічно активних сполук (інкреторні функції).
При ураженні нирок розлади їх екскреторної функції можуть бути обумовлені порушеннями:
1) клубочкоеої (гломерулярної) фільтрації;
2) канальцевої реабсорбції;
3) канальцевої секреції.
Розлади інкреторних функцій нирок можуть виявлятися порушеннями:
1) секреції реніну юкстагломерулярним апаратом нирок, а також ниркових депресорних факторів;
2) вивільнення еритропоетинів та інгібіторів еритропоезу;
3) утворення гормонально активної форми вітаміну D.
32.3. Що таке недостатність нирок? Як її класифікують?
Недостатність нирок - це патологічний стан, для якого характерно порушення сталості внутрішнього середовища організму внаслідок нездатності нирок здійснювати свої гомеостатичні функції.
Ниркову недостатність класифікують у такий спосіб.
I. За клінічним перебігом розрізняють гостру і хронічну ниркову недостатність.
II. Залежно від причин розвитку недостатність нирок може бути преренальною, ренальною, постренальною і аренальною.
III. Залежно від обсягу порушених функцій ниркова недостатність може бути тотальною (порушено всі функції) і парціальною (порушено лише окремі функції).
IV. За механізмами розвитку розрізняють недостатність нирок:
1) пов'язану з первинним ураженням клубочків - гломерулярну;
2) пов'язану з первинним ураженням канальців — тубулярну.
32.4. Які причини гострої ниркової недостатності? Які стадії виділяють у її розвитку?
Гостра ниркова недостатність характеризується швидким виникненням і зна& І чними порушеннями екскреторної функції нирок.
Етіологія гострої недостатності нирок (ГНН) пов'язана з дією внутрішньо- і позаниркових факторів.
Внутрішньониркові фактори ГНН. гострий гломерулонефрит, пієлонефрит, тромбоз і емболія ниркових судин, видалення однієї-єдиної нирки (аренальна ГНН).
Позаниркові фактори ГНН.
а) шок і колапс;
б) гемолітичні та міолітичні стани (переливання несумісної крові, масивне роздавлювання тканин, опіки);
в) зневоднення організму;
г) екзогенна й ендогенна інтоксикації (солями важких металів, оцтовою кислотою, хлороформом, грибною й зміїною отрутами, при токсикозі вагітних, діабетичній комі);
ґ) алергічні стани;
д) порушення виділення сечі внаслідок непрохідності сечоводів або сечівника.
Патогенез ГНН може бути пов'язаний з трьома групами чинників:
1) порушенням кровообігу в нирках (преренальні фактори). Найчастіше ГНН розвивається внаслідок тимчасової ішемії нирок, обумовленої гіповолемією, спазмом аферентних артеріол, ДВЗ-синдромом. Наслідком цього є значне зниження фільг траційного тиску й клубочкової фільтрації, припинення діяльності певної кількості нефронів. Якщо порушення тканинного кровообігу нетривале, то ГНН є оборотним станом (функціональна фаза ГНН). Затяжна ішемія викликає необоротні структурні зміни клубочків і канальців, що відповідає структурній фазі ГНН;
2) прямим ушкодженням структур клубочків і канальців (ренальні фактори). Цим механізмом зумовлюється розвиток ГНН при дії нефротоксичних отрут і деяких інфекційних агентів;
3) порушенням відтоку сечі (постренальні фактори). Ця обставина викликає змен-
Ішення клубочкової фільтрації аж до повного її припинення у зв'язку зі збільшенням тиску первинної сечі в капсулі ниркових клубочків. У клінічному перебігу ГНН виділяють чотири стадії: 1) початкову; 2) ояіго-, анурії; 3) поліурії; 4) видужання.
32.7. Які механізми можуть лежати в основі порушень ниркових функцій?
1. Преренальні — порушення кровопостачання нирок (рис. 146).
2. Ренальнг — порушення функції клубочків (клубочкової фільтрації) і ниркових ка-нальців (канальцевої реабсорбції й секреції). у •'
3. Постренальні - порушення, що виникають на шляху відтоку сечі.
4. Аренальні - порушення, обумовлені відсутністю нирок.
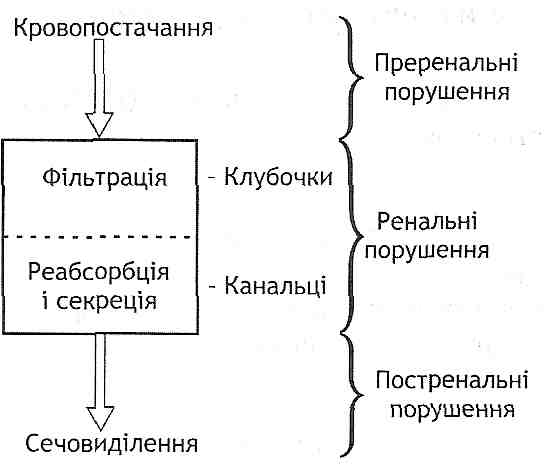
Рис. 146. Механізми порушень ниркових функцій
32.8. У чому-сутність преренальних порушень функцій нирок?
Преренальними називають порушення ниркових функцій, обумовлені розладами кровообігу в нирках.
Інтенсивність ниркового кровообігу в нормі дуже висока (близько 1300 мл/хв. або 25 % хвилинного об'єму крові в стані спокою), що обумовлено його специфічною функцією, тобто участю в здійсненні фільтрації і реабсорбції. Оскільки
![]()
де Qn, - об'ємна швидкість ниркового кровообігу; Ра - тиск на початку, а Рвеп -наприкінці системи перфузії ниркових судин; R - опір ниркових судин; п — в'язкість крові; 1 - довжина судин; г - радіус судин, то зменшення інтенсивності ниркового кровообігу може бути обумовлене:
1) зменшенням артеріального тиску (Р ) нижче 80 мм рт. ст. (не спрацьовує механізм міогенної ауторегуляції - механізм Бейліса). Це спостерігають при всіх видах [ шоку й колапсу;
2) збільшенням венозного тиску (РВСІІ). Причиною цього можуть бути загальні порушення (наприклад, правошлуночкова недостатність серця, що веде до збільшення центрального і периферичного венозного тиску) і місцеві розлади - венозна гіперемія (наприклад, при інтерстиціальному запаленні ниркової тканини);
3) ішемією нирок (зменшенням радіуса судин), що буває при атеросклерозі і артеріальній гіпертензії;
4) збільшенням в'язкості крові (наприклад, при ДВЗ-синдромі).
Усі зазначені порушення спричиняються до зменшення фільтраційного тиску в ниркових клубочках - це виявляє себе зменшенням швидкості клубочкової фільтрації, а отже, ознаками недостатності нирок.
32.9. Чим можуть бути обумовлені порушення клубочкової фільтрації?
Зменшення швидкості клубочкової фільтрації (ШКФ) є основним показником розвитку недостатності нирок. Оскільки
ШКФ = ЕФТ-К.
Ф
де ЕФТ - ефективний фільтраційний тиск; К - коефіцієнт фільтрації, то можна виділити дві групи механізмів порушення клубочкової фільтрації.
I. Зменшення ЕФТ. Оскільки
ЕФТ=Р -(Р + Р),
де Р — гідростатичний тиск у капілярах клубочків; Ро - онкотичний тиск крові; Р — гідростатичний тиск у капсулі клубочків — так званий тканинний тиск, то зменшення ШКФ може бути обумовлено:
1) зменшенням гідростатичного тиску в капілярах клубочків (Рк) унаслідок загальних і місцевих розладів кровообігу (див. запит. 32.8);
2) збільшенням онкотичного тиску крові (Р^, що буває, наприклад, при зневодненні;
3) збільшенням тканинного тиску в нирках (Рт). Причиною цього можуть бути перешкоди відтоку фільтрату або сечі при ушкодженні канальців (закупорка канальців некротичними масами і циліндрами), при інтерстиціальному запаленні (здавлення канальців набряковою рідиною), при порушеннях прохідності сечоводів і сечовивідних шляхів (камені, стриктури, здавлення пухлиною).
II. Зменшення коефіцієнта фільтрації (Кф). Воно може бути обумовлено:
1) зменшенням загальної площі фільтрації, що, у свою чергу, залежить від кількості діючих нефронів;
2) зменшенням проникності стінки клубочкового фільтра, що спостерігається при потовщенні мембрани (наприклад, при діабетичній нефропатії), скле-розуванні клубочків (наслідок гломерулонефриту), забиванні nop фільтра білками (гемоглобіном, міоглобіном відповідно при гемолізі еритроцитів і роздавлюванні м'язової тканини).
32.10. Які фактори можуть викликати збільшення клубочкової фільтрації?
Збільшення фільтрації відбувається під впливом таких чинників.
1. Підвищення гідростатичного тиску у капілярах клубочків, що буває:
а) у разі збільшення об'єму внутрішньосудинного сектора води у зв'язку з прийманням великої кількості рідини, з розсмоктуванням набряків, транссудатів і ексудатів;
б) при зменшенні тонусу привідних;
в) підвищенні тонусу вивідних артеріол.
2. Зниження онкотичного тиску крові, що відбувається у разі перерозподілу білкових фракцій крові у сторону переважання грубодисперсних глобулінів з низьким онкотичним тиском (при гепатиті, цирозі печінки).
32.11. Які механізми можуть лежати в основі порушень функцій ниркових канальців?
1. Ушкодження клітин канальцевого епітелію. Може бути обумовлено ішемією, не-фротропними отрутами, дією фізичних (радіація) і біологічних (інфекція) факторів. При ушкодженні канальців для порушення ниркових функцій мають значення:
а) вихід фільтрату через ушкоджені канальці в інтерстицій, що веде до збільшення тканинного тиску і зменшення клубочкової фільтрації;
б) обтурація канальців некротичними масами і циліндрами, що також зумовлює зменшення ефективного фільтраційного тиску.
2. Зменшення активності ферментів і транспортних білків, що беруть участь у процесах реабсорбції і секреції. Це можуть бути спадково обумовлені дефекти систем транспорту глюкози (ренальна глюкозурія), амінокислот (аміноацидурія), фосфатів (фосфатний нирковий діабет); складні поєднані порушення реабсорбції глюкози, амінокислот, гідрокарбонату і фосфатів (синдром Фанконі). Можливі й набуті розлади транспортних систем, що забезпечують реабсорбцію і секрецію. Наприклад, при отруєнні флоридзином, що пригнічує гексокіназу і глюкозо-6-фосфатазу, розвивається ренальна глюкозурія.
3. Порушення енергозабезпечення. Закономірно виникають при гіпоксії, голодуванні, гіповітамінозах, зменшенні активності ферментів енергетичного обміну і виявляються дефіцитом АТФ. При цьому страждають усі енергозалежні механізми реабсорбції і секреції (первинний і вторинний активний трансмембранний транспорт, ендо- і екзоцитоз).
4. Надлишок речовин, що реабсорбуються. Він зумовлює функціональне перевантаження систем зворотного транспорту. Цей механізм стосується так званих по-рогових речовин, до яких належать глюкоза і гідрокарбонати. Якщо концентрація
цих сполук у крові перевищує пороговий рівень (для глюкози- 10 ммоль/л, для гідрокарбонатів - 27 ммоль/л), то їхній надлишок виводиться із сечею - розвивається глюкозурія (при цукровому діабеті), сеча стає лужною (при алкалозах).
5. Порушення гуморальної регуляції процесів факультативної реабсорбції. Пов'язані зі змінами вмісту в крові альдостерону, передсердного натрійуретично-го гормону, вазопресину, паратирину.
32.12. У чомусутність постренальних порушень ниркових функцій?
Постреішльними називають порушення, що виникають на шляхах виведення сечі. Вони можуть бути обумовлені:
а) обтурацією сечовивідних шляхів (камені) і б) здавленням ззовні — компресією, що виникає як наслідок стриктур сечоводів, аденоми простати й інших пухлин.
Зазначені причини, які перешкоджають відтоку сечі, .викликають підвищення тиску рідини в капсулі клубочків, що призводить до зменшення ефективного фільтраційного тиску і зменшення швидкості клубочкової фільтрації.
Причини та механізми розвитку кількісних і якісних змін складу сечі: олігурія, анурія, поліурія; гіпостенурія, ізостенурія; протеїнурія, гематурія, циліндрурія, лейкоцитурія.
32.13. Які кількісні і якісні зміни сечі можуть наставати при ураженнях нирок?
Кількісні зміни: 1) оліго- і анурія; 2) поліурія; 3) ніктурія; 4) гіпо- та ізостенурія. Якісні зміни: 1) протеїнурія; 2) гематурія; 3) циліндрурія; 4) лейкоцитурія (піурія).
32.14. Що таке оліго- і анурія? У яких випадках виникає олігурія? Які її наслідки?
В умовах нормальної життєдіяльності з організму щодоби має виводитися із сечею 700 мосм осмотично активних речовин. Щоб вивести таку їх кількість при максимально можливій осмоляльності сечі (1000 мосм/кг), необхідно як мінімум 700 мл сечі на добу. Такий добовий об'єм сечі одержав назву облігшпного діурезу, або облігатного об'єму.
Олігурія - це зменшення добового діурезу нижче облігатного об'єму, тобто менше 700 мл/добу.
Анурія — це повна відсутність діурезу.
Причиною олігурії є порушення клубочкової фільтрації. Олігурія виникає тоді, коли під дією преренальних, ренальних і постренальних факторів швидкість клубочкової фільтрації стає нижче 10 мл/хв.
Олігурія призводить до:
1) збільшення об'єму позаклітинної рідини — гіпергідрії (див. розд. 23);
2) накопичення в організмі осмотично активних речовин. Зокрема, розвиваються гі-пернатріємія, гіперкаліємія;
3) накопичення в крові кінцевих продуктів обміну речовин — азотемії.
32.15. Що таке поліурія? Які механізми її розвитку?
Поліурія — це збільшення добового діурезу понад 1,8 л. У людини максимально можливий діурез за умови, що він не осмотичний, дорівнює 25 л/добу, що становить 15 % об'єму профільтрованої води.
Причинами поліурії можуть бути позаниркові (психогенна полідипсія, порушення водно-сольового обміну та його регуляції, наприклад, нецукровий діабет) і ниркові (поліурична стадія гострої і хронічної недостатності нирок) фактори.
Залежно від механізмів розвитку виділяють такі види поліурії.
1. Водний діурез. Обумовлений зменшенням факультативної реабсорбції води. Виникає при водному навантаженні, нецукровому діабеті. Сеча при такій поліурії гіпотонічна, тобто містить мало осмотично активних речовин.
2. Осмотичний діурез (салурез). Пов'язаний зі збільшенням вмісту в сечі нереаб-сорбованих осмотично активних речовин, що спричиняється до вторинного порушення реабсорбції води. Поліурія цього типу розвивається при:
а) порушенні реабсорбції електролітів;
б) збільшенні вмісту в первинній сечі так званих порогових речовин (наприклад, глюкози при цукровому діабеті);
в) дії екзогенних речовин, які погано реабсорбуються (манітол) або порушують реабсорбцію електролітів (салуретики).
В умовах максимального осмотичного діурезу виділення сечі може досягати 40 % величини клубочкової фільтрації.
3. Гіпертензивний діурез. Розвивається при артеріальної гіпертензії, коли збільшується швидкість руху крові в прямих судинах мозкового шару нирок (ці судини йдуть паралельно колінам петлі Генле). При цьому збільшується конвекційний транспорт речовин; саме цей транспорт, а не дифузія стає провідним. Наслідком посилення конвекційного транспорту є "вимивання" натрію, хлору, сечовини з інтерстицію. Це веде до зменшення осмотичного тиску позаклітинної рідини, у результаті зменшується реабсорбція води в нисхідній ділянці петлі Генле і розвивається поліурія.
32.16. Що таке ніктурія? Коли вона виникає?
Ніктурія — це патологічна ознака, сутність якої полягає у переважанні нічної частини діурезу над денною.
У нормі 60-80 % добової кількості сечі виділяється в період з 8 до 20 год., тобто відношення нічного діурезу до денного становить 1:2.
При ніктурії нічна порція сечі може більше ніж удвічі перевищувати денну.
Залежно від причин виділяють:
1) серцеву ніктурію— розвивається при серцевій недостатності. Удень у іворих збільшуються навантаження на серце й приймання води, що веде до застою крові й затримки води в тканинах (набряки). Уночі в горизонтальному положенні поліпшується венозний відтік і зменшується навантаження на серце. Це викликає виділення передсердного натрійуретичного гормону, збільшення діурезу і зменшення набряків;
2) ниркова ніктурія — характерна для ураження нирок. її пояснюють поліпшенням уночі порушеного тканинного кровообігу. У результаті прискорюється рух крові по судинах нирок, розвивається гіпертензивний діурез.
32.17. Що таке гіпо- та ізостенурія?
Гіпостенурія виникає при зменшенні здатності нирок концентрувати сечу. Вона характеризується зменшенням відносної густини сечі до 1012-1006, причому зміни цієї густини протягом доби незначні (рис. 148).
Рис. 148. Добові коливання густини сечі
в нормі, при гіпо- та ізостенурії:
І - норма; II - слабке обмеження; III
- гіпостенурія; IV - ізостенурія
Поєднання гшостенурії з поліурією свідчить про ушкодження канальців при відносно достатній функції клубочків. Якщо гіпостенурія виникає на тлі олігурії, то це ознака ушкодження всіх структур нефронів (канальців і клубочків).
При повній втраті нирками здатності концентрувати і розводити сечу розвивається ізостенурія, при якій відносна густина сечі дорівнює густині фільтрату, тобто 1010, і не міняється протягом доби (монотонний діурез).
Ізостенурія є ознакою дуже важких порушень, при яких ниркові канальці, по суті, перетворюються у звичайні трубки, що проводять фільтрат у ниркові миски.
32.18. Які механізми протеїнурії?
Протеїнурія — це виділення білка із сечею. В основі її розвитку можуть лежати такі механізми:
1) збільшення проникності клубочкового фільтра у зв'язку з ураженням базальної мембрани (клубочкова протеїнурія);
2) зменшення канальцевої реабсорбції білка, що профільтрувався (канальцева про-теїнурія);
3) патологічне надходження білка у просвіт канальців з ушкоджених клітин тубуляр-ного епітелію або з перитубулярної лімфатичної рідини (секреторна протеїнурія).
Протеїнурія може бути селективною, коли в сечі визначають тільки низькомолекулярні білки, і неселективною, для якої характерна поява в сечі як низько-, так і високомолекулярних білків.
За ступенем селективності розрізняють нефротичний тип протеїнурії (у сечі тільки альбуміни або альбуміни+а-глобуліни) і нефритичний тип (у сечі визначаються всі класи білків плазми крові — альбуміни, а-, р- і у-глобуліни).
32.19. Що може бути причиною гематурії, лейкоцитурії, циліндрурії?
Гематурія — поява еритроцитів у сечі. Може бути обумовлена:
1) ушкодженням клубочкового фільтра і надходженням еритроцитів у первинну сечу. При цьому в кінцевій сечі виявляють "вилужені" еритроцити;
2) ушкодженням сечовивідних шляхів.
Лейкоцитурія - поява в сечі лейкоцитів понад 5 у полі зору. Лейкоцитурію, при якій виявляють дуже велику кількість лейкоцитів у сечі, у тому числі й зруйнованих, називають піурією. Основна причина лейкоцитурії — запальні процеси в нирковій тканині і сечовивідних шляхах.
Цііліидрурія — поява в сечі циліндрів. Циліндри являють собою зліпки ниркових канальців. Вони утворюються при ушкодженні епітелію канальців і складаються з осадженого білка і загиблих клітин. Залежно від будови розрізняють гіалінові, зернисті і епітеліальні циліндри.
Синдром гострої ниркової недостатності: визначення поняття, причини та механізми розвитку, клінічні прояви. Нефротичний синдром.
Гостра ниркова недостатність характеризується швидким виникненням і зна& І чними порушеннями екскреторної функції нирок.
Етіологія гострої недостатності нирок (ГНН) пов'язана з дією внутрішньо- і позаниркових факторів.
Внутрішньониркові фактори ГНН. гострий гломерулонефрит, пієлонефрит, тромбоз і емболія ниркових судин, видалення однієї-єдиної нирки (аренальна ГНН).
Позаниркові фактори ГНН.
а) шок і колапс;
б) гемолітичні та міолітичні стани (переливання несумісної крові, масивне роздавлювання тканин, опіки);
в) зневоднення організму;
г) екзогенна й ендогенна інтоксикації (солями важких металів, оцтовою кислотою, хлороформом, грибною й зміїною отрутами, при токсикозі вагітних, діабетичній комі);
ґ) алергічні стани;
д) порушення виділення сечі внаслідок непрохідності сечоводів або сечівника.
Патогенез ГНН може бути пов'язаний з трьома групами чинників:
1) порушенням кровообігу в нирках (преренальні фактори). Найчастіше ГНН розвивається внаслідок тимчасової ішемії нирок, обумовленої гіповолемією, спазмом аферентних артеріол, ДВЗ-синдромом. Наслідком цього є значне зниження фільг траційного тиску й клубочкової фільтрації, припинення діяльності певної кількості нефронів. Якщо порушення тканинного кровообігу нетривале, то ГНН є оборотним станом (функціональна фаза ГНН). Затяжна ішемія викликає необоротні структурні зміни клубочків і канальців, що відповідає структурній фазі ГНН;
2) прямим ушкодженням структур клубочків і канальців (ренальні фактори). Цим механізмом зумовлюється розвиток ГНН при дії нефротоксичних отрут і деяких інфекційних агентів;
3) порушенням відтоку сечі (постренальні фактори). Ця обставина викликає змен-
Ішення клубочкової фільтрації аж до повного її припинення у зв'язку зі збільшенням тиску первинної сечі в капсулі ниркових клубочків. У клінічному перебігу ГНН виділяють чотири стадії: 1) початкову; 2) ояіго-, анурії; 3) поліурії; 4) видужання.
Найбільш характерні і значні порушення спостерігають на стадії оліго-, анурії:
а) різке зменшення (аж до повного припинення) діурезу з розвитком ознак водного отруєння організму - набряк головного мозку, штерстиціальний набряк легень та ін.;
б) тяжкі порушення діяльності системи кровообігу — зменшення скоротливої функції серця, порушення ритму у вигляді екстрасистолії, брадикардії, блокади; артеріальна гіпотензія з наступним переходом у гіпертензію;
в) розлади зовнішнього дихання за типом Куссмауля;
г) значні порушення функцій нервової системи - від головного болю, блювоти до арефлексії, порушень свідомості, судом, коми.
Більша частина хворих з ГНН гине на висоті цієї стадії. При сприятливому перебігу захворювання, а головне - при проведенні ефективних терапевтичних заходів, через 5—10 діб настає перехід у стадію відновлення діурезу й поліурії. Підвищення клубочкової фільтрації спочатку має в своїй основі відновлення цього процесу в не-фронах, що залишилися, а потім (через кілька місяців) збільшення кількості функціонуючих нефронів.
Нефропшчним називають синдром, що виникає при різноманітних ураженнях нирок і виявляється масивною протеїнурією, гіпопротеїнемією, розвитком набряків, гіперліпгдемією.
Нефротичний синдром за походженням поділяють на первинний і вторинний.
Первинний нефротичний синдром не пов'язаний з яким-небудь попереднім захворюванням нирок. Найчастіше в основі його виникнення є наявність генетично обумовлених дефектів обміну речовин (ліпоїдній нефроз) або трансплацентарне перенесення специфічних протиниркових антитіл від матері до плода (уроджений сімейний нефроз).
Вторинний нефротичний синдром обумовлений деякими захворюваннями нирок (гломерулонефрит) або інших органів (нефропатія вагітних, цукровий діабет, амілоїдоз, системний червоний вовчак, сироваткова хвороба, стафілококовий сепсис та ін.). Його спостерігають також при отруєнні солями важких металів, при великих опіках, радіаційному ураженні, при відторгненні ниркового трансплантата, при застосуванні деяких лікарських препаратів (сульфаніламіди, пеніцилін, кортикостероїди).
У патогенезі нефротичного синдрому мають значення два механізми.
I. Імунний механізм. У його основі лежать імунокомплексні реакції (реакції ПІ типу за Кумбсом і Джеллом), які спричиняються до ушкодження базальної мембрани клубочків. Джерелами антигенів можуть бути екзогенні фактори: бактеріальні, вірусні, паразитарні, лікарські, харчові, сполуки важких металів та ін. Ендогенними антигенами можуть бути ДНК, денатуровані нуклеопротеїди, білки пухлинного походження, тиреоглобулін. Антитіла до зазначених антигенів переважно являють собою імуноглобуліни класу Ig M.
Ураження клубочків нирок пов'язують з відкладанням на поверхні або в самій мембрані капілярних судин амілоїду, гліко- і ліпопротеїдів, фібриногену, з активацією гуморальних і клітинних ланок запальної реакції. Внаслідок цього втрачається структурна цілісність базальної мембрани, змінюється її склад і фізико-хімічні властивості, різко підвищується проникність для плазмових білків.
II. Метаболічний механізм і пов'язані з ним фізико-хімічні зміни. Підвищення проникності клубочкового фільтра може бути пов'язане зі зменшенням негативного електричного заряду стінки капілярів клубочків (зумовленого аніонами внутрішньої поверхні судин), зникненням сіалопротеїну, який у нормі тонким шаром покриває ендотелій капілярів і його відростки. У місцях, де втрата аніонів і сіа-лопротеїнів максимальна, накопичуються поліморфноядерні лейкоцити, лізосомні ферменти яких безпосередньо ушкоджують базальну мембрану капілярних судин.
32.28. Які наслідки для організму може мати втрата білків із сечею
при нефротичному синдромі?
Масивна протеїнурія, що її спостерігають при нефротичному синдромі, спричиняється до зменшення вмісту в крові майже всіх функціонально важливих білків. Цим і пояснюють широкий спектр порушень, що виникають в організмі:
Білки, що втрачаються 3 сечею |
Наслідки зменшення вмісту білка в крові |
|
Альбуміни |
|
Ппоонкія, набряки |
Антитромбін III |
|
Схильність до тромбозів і тромбоемболій |
Фактори зсідання крові |
|
Геморагічний діатез |
Компоненти комплементу |
|
Зменшення резистентності до інфекцій |
Імуноглобуліни (Ig) |
|
Те ж саме |
Ліпопротеїди високої густини |
(ЛПВГ) |
Прискорений розвиток атеросклерозу |
Білки, що транспортують мікроелементи |
Дефіцит Fe, Cu, Zn |
|
Білки, що транспортують гормони |
Ендокринні порушення |
|
Синдром хронічної ниркової недостатності: визначення поняття, причини та механізми розвитку, клінічні прояви. Патогенез уремічної коми.
32.5. Які етіологія і патогенез хронічної недостатності нирок?
Етіологічними факторами хронічної недостатності нирок (ХНН) є хронічні прогресуючі захворювання нирок запальної (хронічний гломерулонефрит, хронічний пієлонефрит та ін.), судинної (гіпертонічна хвороба, стеноз ниркової артерії) та метаболічної (діабетичний гломерулосклероз, амілоїдоз, подагра) природи.
У патогенезі ХНН виділяють такі стадії: 1) початкову, 2) ранню поліуричну, 3) пізню олігуричну і 4) термінальну.
ХНН розвивається в результаті одночасного або поступового зменшення маси діючих нефронів (МДН).
Початкові ознаки ХНН з'являються при зменшенні МДН до 50-30 % від вихідної кількості нефронів, клінічно виражена картина розвивається при зниженні МДН до 30—10 % і величини клубочкової фільтрації нижче 20 % (якщо порівнювати з нормою). Дальше зменшення МДН і клубочкової фільтрації (нижче 10 %) веде до розвитку термінальної стадії недостатності нирок —уремії.
Уремічний синдром супроводжує розвиток гострої і хронічної недостатності нирок і розвивається внаслідок порушення клубочкової фільтрації. Перші його ознаки з'являються, коли швидкість клубочкової фільтрації (ІИКФ) стає нижче 50 мл/хв, а виражені клінічні симптоми - при падінні ШКФ нижче 10 мл/хв.
Основу уремічного синдрому становить інтоксикація, обумовлена накопиченням у крові кінцевих продуктів обміну речовин, які в нормі виводяться із сечею. Нині відомо понад 200 речовин, здатних викликати інтоксикацію при недостатності нирок. Найбільше значення мають:
а) сечовина;
б) сечова кислота;
в) похідні гуанідину — креатин, креатинін, метилгуанідин, диметилгуанідин та ін.;
г) ароматичні сполуки - феноли, індол, ароматичні аміни; ґ) аліфатичні аміни;
д) низькомолекулярні пептиди.
Ознакою накопичення зазначених токсичних речовин є азотемія - збільшення вмісту в крові залишкового азоту.
Клінічні прояви уремічного синдрому пов'язані з ураженням практично всіх органів і систем. Однак на перший план виступають ознаки токсичного ураження центральної нервової системи: анорексія, нудота, блювота, психічні порушення, ураження периферичних нервів.
Украй важким проявом інтоксикації є розвиток уремічної коми - повної втрати свідомості зі зникненням рефлексів на зовнішні і внутрішні подразники, пригніченням життєво важливих функцій. Крім інтоксикації, в патогенезі уремічного синдрому мають значення порушення обміну води, електролітів, розлади кислотно-основного стану.
Загальні прояви недостатності ниркових функцій. Патогенез набряків, артеріальної гіпертензії, анемії, порушень гемостазу, кислотно-основного стану, остеодистрофії.
32.7. Які механізми можуть лежати в основі порушень ниркових функцій?
1. Преренальні — порушення кровопостачання нирок (рис. 146).
2. Ренальнг — порушення функції клубочків (клубочкової фільтрації) і ниркових ка-нальців (канальцевої реабсорбції й секреції). у •'
3. Постренальні - порушення, що виникають на шляху відтоку сечі.
4. Аренальні - порушення, обумовлені відсутністю нирок.
32.21. Які механізми можуть обумовлювати розвиток набряків при ураженнях нирок?
Патогенетично розрізняють три типи набряків, що розвиваються при різних ураженнях нирок.
1. Набряки при гострій і хронічній недостатності нирок. Основний механізм їх розвитку — гідростатичний (гіперволемічний) (див. розд. 23). Зменшення швидкості клубочкової фільтрації, характерне для ниркової недостатності, призводить до затримки натрію й води в організмі (позитивний водний баланс) і, як наслідок, до гіперволемії. Остання, будучи причиною збільшення гідростатичного тиску в капілярах, викликає розвиток набряків за механізмом Старлінга.
2. Нефротичні набряки. Основний механізм їх розвитку — онкотичний (гіпопроте-гнемічний). Порушення клубочкового фільтра при нефрозі викликають масивну протеїнурію, у результаті якої розвивається гіпопротеїнемія і падає онкотичний тиск крові. Це, у свою чергу, за механізмом Старлінга викликає перехід води з судин у тканини - розвиваються набряки.
3. Нефритичні набряки. Розвиваються при гострому і хронічному гломерулонефриті. Патогенез цих набряків складний і в своїй основі має такі механізми:
а) запалення клубочків → застій крові в судинах нирок → гіпоксія юкстагло-мерулярного апарату → активація ренін-ангіотензинної системи → секреція альдостерону → затримка натрію в організмі і підвищення осмотичного тиску крові → секреція антидіуретичного гормону → затримка води → гі-перволемія → набряки;
б) запалення клубочків → порушення ниркового кровообігу → зменшення швидкості клубочкової фільтрації → затримка натрію й води в організмі → гіперволемія → набряки;
в) запалення клубочків → збільшення проникності ниркового фільтра →про-теїнурія → гіпопротеїнемія → набряки.
32.22. Які порушення кислотно-основного стану можуть розвиватися при ураженнях нирок?
Найчастіше розвивається негазовий ацидоз. Залежно від сутності патологічних процесів у нирках можливі три його варіанти.
1. Клубочковий ацидоз (нирковий азотемічний ацидоз). Виникає внаслідок розвитку ниркової недостатності при зменшенні швидкості клубочкової фільтрації ниж-
че 25 мл/хв. Він є метаболічним ацидозом зі збільшеною аніонною різницею (див. розд. 25). Його розвиток обумовлений накопиченням іонів водню, що утворюються ендогенно, в основному у формі сірчаної, фосфорної, сечової та інших кислот.
2. Проксимальний нирковий канальцевий ацидоз. Є наслідком первинних порушень реабсорбції гідрокарбонату в проксимальних звивистих канальцях не-фронів. Його відносять до видільних ацидозів з нормальною аніонною різницею (гіперхлоремічний).
3. Дистальний нирковий канальцевий ацидоз. Обумовлений первинними порушеннями процесів ацидогенезу в дистальних звивистих канальцях, де відбувається відтитровування буферів (збереження гідрокарбонату) і підкислення (аци-дифікація) сечі. Є метаболічним ацидозом з нормальною аніонною різницею (гіперхлоремічний). Виявляється нездатністю витримувати ендогенне навантаження іонами водню. При цьому рН сечі не досягає значень нижче 6.
Зміни функції нирок іноді можуть спричинятися до розвитку негазового гіпо-каліємічного алкалозу. Це, зокрема, буває при гіперальдостеронізмі. Альдостерон, посилюючи секрецію іонів калію в дистальних звивистих канальцях, викликає гіпо-каліємію, яка, у свою чергу, обумовлює виникнення алкалозу (див. розд. 25).
32.23. Які порушення фосфорно-кальцієвого обміну характерні для недостатності нирок? Який їх патогенез?
Порушення фосфорно-кальцієвого обміну при недостатності нирок виявляються розвитком ниркової остеодистрофії (остеопатії).
Ниркова остеодистрофія — це комплекс дистрофічних порушень у кістках, що виникають унаслідок розладів фосфорно-кальцієвого обміну при ураженнях нирок. Клінічно вона виявляє себе двома групами змін.
I. Дистрофічні зміни в кістках:
а) резорбція кісткової тканини (фіброзно-кістозний остеїт) — у кістках утворюються порожнини, які заповнюються фіброзною тканиною. Є наслідком гіперпаратиреозу;
б) остеомаляція - розм'якшення кісток, їх деформація, болі в кістках. У дітей порушується окостеніння хрящів. Є наслідком гіпокальціємії;
в) остеопороз — зменшення щільності кісткової тканини без деформації кісток;
г) остеосклероз — збільшення щільності кісткової тканини.
II. Обвапнеїшя м'яких тканин - кальцифікація (див. розд. 24).
У патогенезі ниркової остеодистрофії мають значення такі механізми:
а) хронічна недостатність нирок (ХНН) → зменшення екскреції фосфатів → гіперфосфатемія → гіпокальціємія (див. розд. 24) → збільшення продукції паратирину → резорбція кісткової тканини, її демінералізація;
б) ХНН → порушення перетворення 25-оксивітаміну D у його гормонально активну форму- 1,25-діоксивітамін D → зменшення всмоктування іонів Са2+ у кишках → гіпокальціємія → див. вище;
в) ХНН → клубочковий ацидоз → обмін іонів Са2+ і Na+ на іони Н+ крові (буферна функція кісткової тканини) → демінералізація кісток.
32.24. Які види артеріальної гіпертензії можуть виникати при ураженнях нирок? Який їх патогенез?
Артеріальна гіпертензія є одним з проявів хронічної недостатності нирок (ХНН). Виділяють такі її види.
1. Гіпертензія, залежна від об'єму. Цей вид буває у 80-90 % хворих з ХНН. Гї патогенез пояснюють таким чином: ХНН → зменшення швидкості клубочкової фільтрації → порушення екскреції натрію і води → гіперволемія → збільшення хвилинного об'єму серця → збільшення артеріального тиску.
2. Гіпертензія з високим рівнем реніну (високоренінова). Цей вид виявляють в.І 5-10 % хворих з ХНН. Основний її механізм: ураження нирок → порушення ниркового кровообігу → гіпоксія юкстагломерулярного апарату → вивільнення реніну → активація ренін-ангіотензинної системи → далі див. розд. 28.
3. Гіпертензія, не залежна від об'єму. Буває в 5-10 % хворих з ХНН. Характеризується нормальним об'ємом циркулюючої крові й нормальним рівнем реніну в крові. Вважають, що розвиток цього виду гіпертензії пов'язаний з порушенням депресорних функцій нирок, а саме — зі зменшенням утворення ниркових простагландинів, зокрема ПГ Е2, зменшенням активності ниркової калікреїн-кінінової системи, пригніченням утворення нейтрального ліпіду, що має гіпотензивну дію.
32.25. Які механізми обумовлюють розвиток анемії, що супроводжує недостатність нирок?
Ознаки анемії у хворих з хронічною недостатністю нирок з'являються при зменшенні швидкості клубочкової фільтрації нижче 40 мл/хв. У її патогенезі мають значення такі механізми.
1. Пригнічення еритропоезу:
а) порушення регуляції еритропоезу, обумовлене пригніченням утворення ниркових еритропоетинів і посиленим продукуванням інгібіторів кровотворення;
б) ушкодження кровотворних клітин уремічними токсинами;
в) дефіцит заліза, що розвивається як наслідок хронічних крововтрат при захворюваннях нирок і втрат трансферину в результаті протеїнурії.
2. Крововтрати. Можуть бути пов'язані з гематурією, з частим утворенням виразок у шлунку і кишках хворих з ураженням нирок, з розвитком геморагічного діатезу, і
3. Гемоліз еритроцитів, обумовлений уремічними токсинами.
32.26. Якими порушеннями гемостазу може виявляти себе недостатність нирок?
І. ТрОхМбофілічні порушення- гіиеркоагуляція. В основі їх розвитку можуть бути:
а) зменшення активності фібринолітичної системи внаслідок дефіциту урокі-нази — активатора плазміногену;
б) зменшення активності антикоагулянтної системи, що пов'язано з порушеннями синтезу і депонування гепарину в нирках;
в) втрата із сечею внаслідок протеїнурії антитромбіну III— основного природного антикоагулянта. її. Геморагічний діатез. Може бути обумовлений розладами як судинно-тромбо-цитарного гемостазу (тромбоцитопенія внаслідок мієлотоксичної дії уремічних токсинів), так і коагуляційного (втрата із сечею факторів зсідання крові при протеїнурії).
Гломерулонефрит: визначення поняття, принципи класифікації, експериментальні моделі. Етіологія, патогенез дифузного гломерулонефрит
Гломерулонефрит - це двостороннє дифузне захворювання нирок алергічної природи.
Розрізняють гострий і хронічний гломерулонефрит.
Для гострого гломерулонефриту характерні бурхливий початок, олігурія, протеїнурія, азотемія, артеріальна гіпертензія, набряки, гематурія, порушення з боку центральної нервової системи.
Гострий гломерулонефрит виникає під час (або після) якої-небудь інфекції, частіше стрептококової природи. Вважають, що гемолітичний стрептокок групи А (типи 4,12) є специфічним "нефритогенним" штамом. Певну роль відіграють й інші інфекції, у тому числі вірусні, паразитарні. Встановлено етіологічну роль охолодження, дифузних уражень сполучної тканини (системний червоний вовчак, ревматоїдний артрит, вузликовий періартеріїт), опікової хвороби, попередньої вакцинації або використання з лікувальною метою гетерологічних сироваток.
Виділяють два основних патогенетичних варіанти гострого гломерулонефриту.
1. Ураження базальної мембрани клубочків нефронів антитілами проти її антигенів - нефротоксичний гломерулонефрит (має стрімкий перебіг, швидко прогресує). Носієм антигенних властивостей базальної мембрани є глікопротеїд.
2. Розвиток запального процесу в клубочках унаслідок фіксації на базальній мембрані та всередині її імунних комплексів - імунокомплексний гломерулонефрит.
У розвитку такого типу хвороби можуть брати участь антигени як екзогенного (інфекційної чи неінфекційної природи), так і ендогенного (білок тканин, ДНК) походження. Антитіла (Ig G, Ig М) прямо в крові вступають у взаємодію із зазначени-
ми антигенами, потім у вигляді імунних комплексів (антиген-антитіло-комплемент) надходять у клубочки, відкладаючись на їхній базальній мембрані. Реалізація ушкоджувальної дії імунних комплексів, як і нефротоксичних антитіл, відбувається через розвиток імунного запалення (див. розд. 10).
До імунокомплексних відносять гломерулонефрити, що розвиваються після стрептококової інфекції, при системному червоному вовчаку, сироватковій хворобі та ін. Більшість випадків гломерулонефриту (не менше ніж 80 %) є імунокомплексними.
32.30. Які існують експериментальні моделі гострого гломерулонефриту?
1. Введення тваринам нефротоксичної сироватки, тобто сироватки, що містить антитіла проти антигенів ниркової тканини.
Серед моделей цієї групи найчастіше використовують модель Ліндемана і модель Масугі.
Модель Ліндемана. Кролям внутрішньовенно вводять нефротоксичну сироватку, отриману від морської свинки, попередньо імунізованої суспензією нирки кроля. При цьому відбувається фіксація протиниркових антитіл на базальних мембранах клубочків з наступним зв'язуванням комплементу і розвитком ушкодження. Модель Масугі. Кролям внутрішньовенно вводять нефротоксичну сироватку качок, імунізованих тканиною нирок кролів. Особливість цієї моделі полягає в тому, що антитіла птахів фіксуються на базальній мембрані клубочків, але не можуть зв'язувати комплемент ссавців. Це призводить до утворення власних кролячих антитіл проти фіксованих качиних (для цього треба 6—8 діб). Останні в цьому випадку виступають антигенами. Кролячі антитіла, з одного боку, взаємодіють з фіксованими на базальній мембрані качиними, з другого - зв'язують комплемент, активація якого і призводить до ушкодження клубочків.
2. Охолодження нирки — заморожування хлороформом (Герцен). При цьому в крові дослідних тварин (кролів) з'являються специфічні протиниркові антитіла і зазнає уражень друга інтактна нирка. Зазначена модель доводить роль аутоімунних механізмів у розвитку так званого "окопного" нефриту (нефриту воєнного часу).
3. Введення в черевну порожнину кролів клітинної суспензії ниркової тканини і культури стрептококів (подружжя Ковелті).
4. Імунізація овець базальними мембранами клубочків нефронів нирки людини в повному ад'юванті Фрейнда (Стеблей).
5. Імунізація щурів суспензією гомологічної або аутологічної нирки з повним ад'ю-вантом Фрейнда (Хеймен).
6. Виведення чистих ліній тварин (наприклад, новозеландські миші лінії NZB/BL), у яких спонтанно виникають аутоімунні хвороби, у тому числі і гломерулонефрит
32.31. Чим може виявлятися хронічний гломерулонефрит? Які його етіологія й патогенез?
Хронічний гломерулонефрит являє собою тривале прогресуюче дифузне двостороннє ураження нирок запальної природи, неоднорідне за походженням, клінічними проявами і патогенезом.
Залежно від клінічних проявів виділяють такі його форми.
1. Латентна форма виявляє себе ізольованим сечовим синдромом — помірною про-теїнурією, гематурією. У частини хворих спостерігають набряки і транзиторну артеріальну гіпертензію.
2. Гіпертонічна форма характеризується стійким підвищенням артеріального тиску, набряками, гематурією, протеїнурією, циліндрурією і лейкоцитурією.
3. Нефротична форма, яку супроводжують набряковий синдром, виражена протеї-нурія і циліндрурія, зміни крові (гіпопротеїнемія, гіперліпідемія).
4. Змішана, або нефротично-гіпертонічна форма, для якої характерні набряк і гіпертензія, але на відміну від нефротичної форми нема характерних змін крові.
Хронічний гломерулонефрит може бути наслідок гострого, але частіше розвивається первинно. Залежно від причин виділяють такі його форми:
1) інфекційного походження (постстрептококовий, при затяжному септичному ендокардиті, малярії, сифілісі, туберкульозі та інших інфекціях);
2) неінфекційні (сироватковий, вакцинний, медикаментозний, травматичний, при отруєннях, охолодженні, тромбозі ниркових вен);
3) зумовлені дифузними захворюваннями сполучної тканини (ревматоїдний артрит, системний червоний вовчак, геморагічний васкуліт та ін.);
4) особливі (після еклампсії, радіаційний, спадковий та ін.).
Загальновизнаною є імунологічна концепція розвитку хронічного гломерулонефриту. Поряд з нефротоксичними та імунокомплексними механізмами (див. запит. 32.29) певне значення в його патогенезі має гіперчутливість уповільненого типу.
Типові порушення діяльності ендокринних залоз у дітей, їх причини та механізми розвитку. Порушення прямих та зворотних регуляторних зв'язків в патогенезі дисрегуляторних ендокринопатій.
I. Гіперфункція ендокринних залоз (ендокринна гіперфункція).
II. Гіпофункція ендокринних залоз (ендокринна гіпофункція).
III. Дисфункція ендокринних залоз (ендокринна дисфункція) — різноспрямовані зміни функції ендокринних залоз.
33.5. Які виділяють патогенетичні варіанти порушень ендокринних функцій?
Рис. 149. Патогенетичні варіанти порушень ендокринних функцій
I. Дисрегуляторні порушення - розлади регуляції ендокринних залоз.
II. Залозисті порушення — розлади біосинтезу гормонів та їх секреції.
III. Периферичні порушення — розлади транспорту, рецепції і метаболізму гормонів (рис. 149).
33.6. Як здійснюється регуляція ендокринних функцій? Чим можуть бути обумовлені їх дисрегуляторні порушення?
Регуляція діяльності ендокринних залоз може здійснюватися за допомогою чотирьох механізмів (рис. 150).
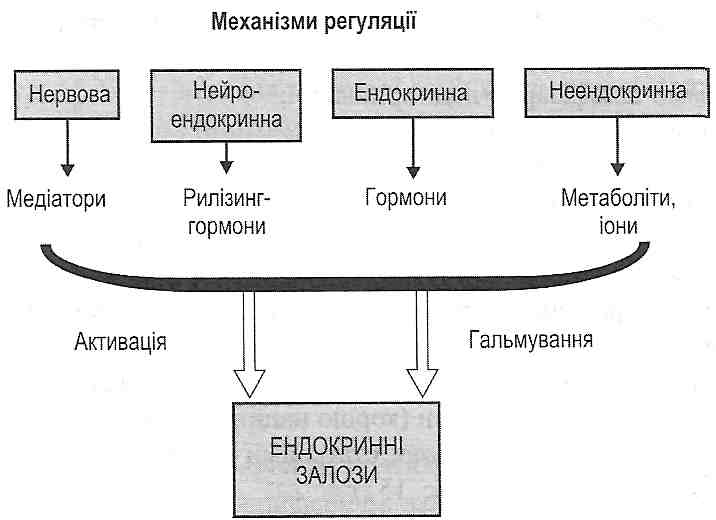
Рис. 150. Механізми регуляції ендокринних залоз
1. Нервова (імпульсно-медіаторна) регуляція. За допомогою прямих нервових впливів регулюється діяльність:
а) мозкового шару надниркових залоз;
б) нейроендокринних структур гіпоталамуса;
в) епіфіза.
2. Нейроендокринна (гіпоталамічна) регуляція. Здійснюється нейроендокринними клітинами гіпоталамуса, що трансформують нервові імпульси у специфічний ендокринний процес. Сутність цього процесу полягає в утворенні і секреції в систему портальних судин гіпофіза рилізинг-гормонів, що регулюють діяльність аде-ногіпофіза.
3. Ендокринна регуляція. Сутність її полягає в безпосередньому впливі одних гормонів на синтез і секрецію інших. Прикладом цього механізму регуляції є вплив тропних гормонів аденогіпофіза на діяльність кори надниркових залоз, щитоподібної залози, статевих залоз.
4. Неендокринна гуморальна регуляція. Здійснюється неспецифічними гуморальними факторами, зокрема метаболітами, іонами. Так, концентрація глюкози в крові прямо впливає на синтез і секрецію інсуліну і глюкагону; склад і рівень амінокислот — на утворення соматотропного гормону; вміст іонів калію — на виділення в кров альдостерону; концентрація кальцію - на секрецію паратирину і кальцитоніну. Розвиток дисрегуляторних порушень ендокринних функцій може бути пов'язаний з розладами всіх чотирьох механізмів регуляції. В одних випадках, при активацій зазначених механізмів розвивається ендокринна гіперфункція, в інших, при пригнд чені регуляторних впливів, - ендокринна гіпофункція.
33.7. Які принципи лежать в основі регуляції ендокринних функцій? Як вони можуть порушуватися?
І. Принцип прямих зв'язків. Лежить в основі діяльності таких ендокринних функціональних систем:
а) гіпоталамо-гіпофізарно-наднирникової;
б) гіпоталамо-гіпофізарно-тиреоїдної;
в) гіпоталамо-гіпофізарно-гонадальної.
Цей принцип реалізується через здійснення трьох послідовних етапів:
1) сприйняття гіпоталамусом вищих нервових регуляторних сигналів і секреція відповідних рилізинг-гормонів;
2) у відповідь на дію рилізинг-гормонів секреція аденогіпофізом відповідних троп-них гормонів;
3) у відповідь на дію тропних гормонів утворення і виділення периферичними залозами (корою надниркових залоз, щитоподібною залозою, статевими залозами) ефекторних гормонів (рис. 151).
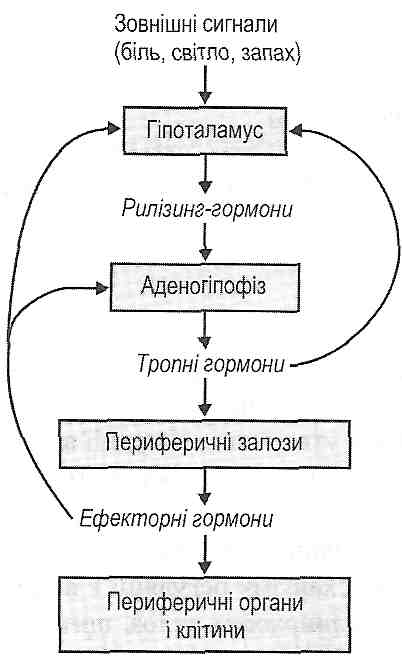
Рис. 151. Прямі і зворотні
зв 'язки в системі гіпоталамус-
аденогіпофіз-периферичні залози
Порушення прямих зв'язків у регуляції ендокринних функцій можуть бути обумовлені розладами синтезу і секреції:
а) рилізинг-гормонів;
б) тропних гормонів;
в) ефекторних гормонів.
II. Принцип зворотних зв'язків. Розрізняють зворотні негативні зв'язки в системах гіпоталамус-аденогіпофіз-периферичні залози й короткі парагі-пофізарні зворотні зв'язки.
Сутність негативних зворотних зв'язків у системах гіпоталамус-аденогіпофіз-периферич-ні залози полягає в тому, що утворювані гормони пригнічують діяльність структур, які здійснюють попередні етапи регуляції. Внаслідок цього посилення секреції ефекторного гормону через певні ланки призводить до зменшення його утворення і надходження в кров, і навпаки, зменшення вмісту гормону в крові викликає підвищення інтенсивності його утворення і секреції.
Порушення негативних зворотних зв'язків у системах гіпоталамус-аденогіпофіз-периферичні залози мають велике значення в розвитку ендокринної патології. Це положення можна проілюструвати схематично такими прикладами.
1. Система гіпоталамус-аденогіпофіз-щитоподібна залоза. Дефіцит йоду в продуктах харчування і питній воді → зменшення утворення тиреоїдних гормонів → стимуляція гіпоталамуса і аденогіпофіза → збільшення утворення тиреолі-берину і тиреотропного гормону → гіпертрофія щитоподібної залози → ендемічний зоб.
2. Система гіпоталамус—аденогіпофіз—кора надниркових залоз. Тривале застосування з лікувальною метою глюкокортикоїдних препаратів → пригнічення діяльності відповідних структур гіпоталамуса і аденогіпофіза → зменшення утворення кор-тиколіберину і АКТГ → атрофія пучкової зони кори надниркових залоз → зменшення утворення власних глюкокортикоїдів → при різкій відміні глюкокортикоїдних препаратів розвивається синдром гострої недостатності кори надниркових залоз (синдром відміни).
3. Система гіпоталамус-аденогіпофіз-статеві залози. Тривале застосування (наприклад, спортсменами-важкоатлетами) анаболічних стероїдів — похідних чоловічих статевих гормонів → пригнічення діяльності відповідних структур гіпоталамуса і аденогіпофіза → зменшення утворення гонадотропних гормонів → атрофія клітин сім'яників, що продукують чоловічі статеві гормони → розвиток імпотенції і безплідності.
Короткі парагіпофізарні зворотні зв'язки (рис. 152) здійснюються поза системою гіпоталамус-аденогіпофіз-периферичні залози. Вони відіграють провідну роль у регуляції діяльності прищитоподібних залоз, β-клітин острівців підшлункової залози. Ці зв'язки можна описати такими схемами.
Прищитоподібні залози', зменшення концентрації іонів кальцію в плазмі крові → збільшення продукції паратирину → збільшення вмісту іонів кальцію → пригнічення секреції паратирину.
β-клітини острівців підшлункової залози: збільшення концентрації глюкози в крові → активація утворення і секреції інсуліну → зменшення вмісту глюкози в крові → пригнічення секреції інсуліну.
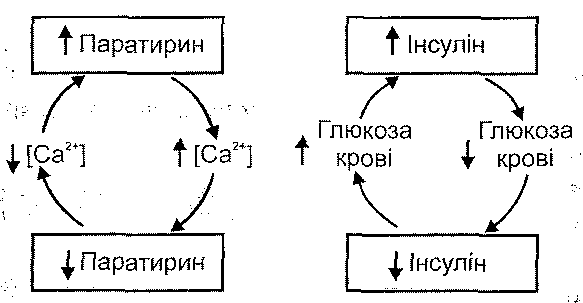
Рис. 152. Короткі парагіпофізарні зворотні зв'язки
Залозисті ендокринопатії. Причини та механізми порушень біосинтезу, депонування та секреції гормонів.
33.8. Чим можуть бути обумовлені власне залозисті порушення ендокринних функцій?
I. Змінами кількості функціонально активних ендокринних клітин:
а) зменшенням їх кількості (видалення залози або її частин, ушкодження, некрози), що призводить до розвитку ендокринної гіпофункції;
б) збільшенням їх кількості (доброякісні і злоякісні пухлини залозистого епітелію), що супроводжується ознаками відповідної ендокринної гіперфункції.
II. Якісними змінами в ендокринних клітинах:
а) розладами біосинтезу гормонів;
б) порушеннями процесів їх секреції.
33.9. Назвіть основні причини порушень біосинтезу гормонів.
Причини порушення синтезу білково-пептидних гормонів:
1) порушення транскрипції;
2) порушення трансляції;
3) дефіцит необхідних амінокислот;
4) дефіцит АТФ;
5) порушення посттрансляційної модифікації і активації.
Причини розладів синтезу стероїдних гормонів:
1) порушення надходження в клітини, синтезу і депонування холестеролу - вихідної речовини для синтезу стероїдів;
2) набуті або спадково обумовлені дефекти ферментів, які беруть участь у реакціях біосинтезу стероїдних гормонів;
3) дефіцит кисню (гіпоксія), необхідного для реакцій гідроксилювання стероїдів;
4) дефіцит відновленого НАДФ (НАДФН) - основного джерела електронів і протонів у реакціях гідроксилювання стероїдів.
Причини порушення синтезу гормонів — похідних амінокислот:
1) дефіцит вихідних амінокислот (тирозину, триптофану);
2) дефіцит мікроелементів (йоду для утворення тиреоїдних гормонів);
3) набуті або спадково обумовлені дефекти ферментів синтезу цих гормонів;
4) дефіцит АТФ.
33.10. Як здійснюється секреція гормонів? Що може обумовлювати її порушення?
Існує три механізми секреції гормонів ендокринними клітинами:
1) вивільнення гормону із клітинних секреторних гранул (секреція білково-пептидних гормонів і катехоламінів);
2) вивільнення гормону з білковозв 'язаної форми (секреція тиреоїдних гормонів);
3) відносно вільна дифузія гормонів через клітинні мембрани (секреція стероїдних гормонів).
В основі розладів секреції гормонів можуть лежати такі механізми: а) порушення депонування гормонів. При цьому страждає утворення комплексів гормонів з речовинами — факторами депонування (білками-нейрофізинами для вазопреси-
ну і окситоцину, АТФ - для катехоламінів, цинком - для інсуліну). Як наслідок, гормони не здатні накопичуватися в необхідних кількостях у секреторних гранулах;
б) порушення передачі сигналів, що стимулюють секрецію. Вони часто пов'язані зі зменшенням утворення в клітинах або надходження вторинних посередників (цАМФ, іонів Са2+);
в) ураження контрактильних елементів (мікрофіламентів, мікротрубочок), що беруть участь у процесах екзо- і ендоцитозу. Ці процеси становлять основу секреції білково-пептидних гормонів (екзоцитоз) і тиреоїдних гормонів (ендоцитоз);
г) дефіцит АТФ, унаслідок чого порушується забезпечення енергозалежних процесів транспорту гормонів
Периферичні розлади ендокринної функції. Розлади транспорту та інактивації гормонів. Порушення рецепції гормонів. Механізми гормональної резистентності.
33.11. Які порушення можуть лежати в основі розвитку периферичних розладів ендокринних функцій?
1. Порушення транспорту гормонів в організмі.
2. Розлади метаболічної інактивації гормонів.
3. Порушення взаємодії гормонів з периферичними клітинами-мішенями - патологія циторецепції гормонів.
33.12. Як здійснюється транспорт гормонів в організмі? Які порушення ендокринних функцій можуть бути пов 'язані з його розладами?
Існує чотири форми транспорту гормонів в організмі.
1. Транспорт вільного гормону (розчиненого у воді). Саме в такій формі гормон виявляє свою біологічну активність, отже, від концентрації вільної форми гормону залежать його функціональні, структурні й біохімічні ефекти. У нормі вміст вільних гормонів у крові не перевищує 10 % від загальної їх кількості.
2. Комплекси гормонів зі специфічними транспортними білками плазми крові. Вміст цієї транспортної форми в крові становить 80 % і більше від сумарної концентрації даного гормону.
3. Неспецифічні комплекси гормонів з білками плазми крові (альбумінами, а-глобу-лінами).
4. Адсорбція гормонів на поверхні формених елементів крові (еритроцитів, лімфоцитів, моноцитів).
Порушення транспорту гормонів в організмі можуть виявлятися двома типами розладів ендокринної функції.
З одного боку, при збільшенні зв'язування гормону зменшується вміст його вільної форми, а отже, з'являються ознаки відповідної ендокринної гіпофункції. З другого боку, зменшення зв'язування гормону викликає збільшення в крові концентрації його вільної форми, що виявляється ознаками відповідної ендокринної гіперфункції.
33.13. Як здійснюється метаболічна інактивація гормонів? Які порушення ендокринної функції можуть бути пов'язані з розладами метаболізму гормонів ?
Руйнування білково-пептидних гормонів швидко відбувається в печінці під дією ферментів пептидаз.
Інактивація стероїдних гормонів здійснюється в печінці, кишках, нирках - практично у всіх органах і тканинах, за винятком тиміко-лімфоцитарної системи. У реакціях перетворення стероїдів беруть участь НАДФН-залежні ферменти. Інактивовані форми стероїдних гормонів, що утворилися в різних органах, надходять у печінку, де відбувається їх кон'югація із сірчаною і глюкуроновою кислотами з подальшим виведенням з організму у складі сечі і калу.
Інактивація катехоламінів може відбуватися трьома шляхами:
1) перетворення, обумовлені моноамінооксидазою (МАО-шлях);
2) вплив катехолоксиметилтрансферази (КОМТ-шлях);
3) хіноїдне окиснення з утворенням адренохрому.
Метаболічні перетворення тиреоїдних гормонів, що відбуваються переважно в печінці, охоплюють:
1) реакції дейодування;
2) окисне дезамінування і декарбоксилювання залишків аланіну;
3) кон'югацію із сірчаною і глюкуроновою кислотами.
У людини 65—95 % інактивованих метаболітів усіх гормонів виводиться з організму із сечею.
Порушення метаболічних перетворень гормонів можуть обумовлювати розвиток периферичних розладів ендокринної функції. Так, при уповільненні інактивації гормонів збільшується їх вміст у крові, що виявляється ознаками відповідної ендокринної гіперфункції. І навпаки, прискорене перетворення гормонів у неактивні форми супроводжується розвитком ендокринної гіпофункції.
33.14. Які можливі порушення взаємодії гормонів з периферичними клітинами?
Порушення взаємодії гормонів з периферичними клітинами-мішенями, як правило, обумовлене патологією клітинних рецепторів (цumopeuenmopiв). Можливі такі варіанти порушень.
I. Зменшення кількості рецепторів або їхньої спорідненості до гормону (десенсити-зація). При цьому, незважаючи на те що концентрація гормону в крові нормальна або навіть підвищена, розвиваються ознаки ендокринної гіпофункції.
II. Збільшення кількості рецепторів до гормону (сенситизація). Як правило, супроводжується розвитком елементів відповідної ендокринної гіперфункції.
33.15. Як відбувається реалізація біологічної дії гормонів на клітини?
Вплив гормонів на клітини-мішені здійснюється через їхню дію на специфічні
білки, що одержали назву циторецепшорів.
Розрізняють два принципово різних типи циторецепції гормонів.
І. Внутрішньоклітинний тип циторецепції. Лежить в основі механізму дії стероїдних і тирео'їдних гормонів. Пов'язаний з вільним проходженням гормону через/ плазматичну мембрану в клітину, у цитоплазмі якої відбувається взаємодія гормону із внутрішньоклітинними білками-рецепторами.
Ефекторною структурою клітини, на яку впливає утворений комплекс, є ядро, а основним біологічним ефектом — зміни інтенсивності процесів транскрипції, а отже, і синтезу клітинних білків.
II. Мембранний тип циторсцепції. Є основним механізмом дії білково-пептидних гормонів і катехоламінів. При цьому гормони не проникають усередину клітини, а зв'язуються з білками-рецепторами на поверхні плазматичної мембрани. Далі передача регуляторного сигналу з поверхні клітини до її ефекторних структур обумовлена появою в цитоплазмі так званих вторинних посередників, або ме-сенджерів. У результаті виникають швидкі біохімічні ефекти, пов'язані з активацією вже синтезованих ферментів чи інших білків. Серед відомих сьогодні вторинних посередників (месенджерів) такі сполуки:
1) циклічні нуклеотиди — цАМФ, цГМФ;
2) іониСа2+;
3) фосфоліпідні месенджери — діацилгліцерол (ДАГ) та інозитолтрифосфат (ІФ3).
Патологія нейроендокринної системи. Причини виникнення та механізми розвитку синдромів надлишку та нестачі гіпофізарних гормонів, їх загальна характеристика.
33.16. Яке значення гіпоталамуса в регуляції ендокринних функцій?
Гіпоталамус є відділом центральної нервової системи, де відбувається інтегрування нервових і ендокринних механізмів регуляції. Це пов'язано з тим, що нейрони гіпоталамуса, об'єднані в окремі ядра, є особливими нейронами — нейроендокринними клітинами, здатними синтезувати і вивільняти гормони.
Гіпоталамус анатомічно і функціонально пов'язаний з адено- і нейрогіпофізом. Тому виділяють дві функціональні системи: гіпоталамо-аденогіпофізарну і гіпотала-мо-нейрогіпофізарну.
Діяльність гіпоталамо-аденогіпофізарної системи пов'язана з утворенням у гіпоталамусі гіпофізотропних гормонів - рилізипг-гормоиів. Залежно від функціональних ефектів (активація або пригнічення функції аденогіпофіза) їх поділяють на дві групи: ліберини і статини. До ліберинів (активаторів секреторної функції аденогіпофіза) відносять, зокрема, тиреоліберин, соматоліберин, кортиколіберин, гонадоліберин, пролактоліберин, меланоліберин. Статинами, що пригнічують функції аденогіпофіза, є соматостатин, пролактостатин, меланостатин.
В основі функціонування гіпоталамо-нейрогіпофізарної системи лежить утворення в супраоптичному і паравентрикулярних ядрах гіпоталамуса двох гормонів — вазопресину і окситоцину.
33.17. Які причини можуть викликати порушення функції гіпоталамо-аденогіпофізарної системи?
1. Патогенна дія факторів зовнішнього і внутрішнього середовища, які у нормі регулюють стан гіпоталамо-аденогіпофізарної системи (негативні емоції, біль, психічні порушення та ін.).
2. Ураження відділів центральної нервової системи, які чинять регуляторний вплив на гіпоталамус, - вищих центрів кори великих півкуль головного мозку, структур лімбічної системи, ретикулярної формації.
3. Ураження гіпоталамуса.
4. Ураження аденогіпофіза.
33.18. Які механізми можуть лежати в основі гіпер- і гіпофункції гіпоталамо-аденогіпофізарної системи?
I. Порушення центральної регуляції нейроендокринних зон гіпоталамуса. При збільшенні активуючих і зменшенні гальмівних впливів розвивається гіперфункція гіпоталамо-аденогіпофізарної системи. Зменшення активуючої дії і збільшення гальмівних впливів, навпаки, викликають гіпофункцію цієї системи.
II. Порушення утворення і виділення рилізинг-гормонів клітинами гіпоталамуса. При цьому гіперфункція гіпоталамо-аденогіпофізарної системи розвивається при збільшенні секреції ліберинів і зменшенні утворення статинів, а гіпофункція - при зменшенні виділення ліберинів і збільшенні секреції статинів.
Ш. Порушення утворення і секреції гормонів аденогіпофіза. Залежно від спрямованості цих порушень можуть розвиватися ендокринна гіпер- або гіпофункція.
33.19. Які гормони утворюються в аденогіпофізі? Які біологічне ефекти вони мають?
1. Соматотропний гормон (СТГ, соматотропін, гормон росту). Утворюється ацидофільними клітинами аденогіпофіза. Має ростову і метаболічну активність. Ростова активність пов'язана з дією СТГ на рецептори гепатоцитів, клітин ниркового епітелію та ін., унаслідок чого вивільняються тканинні фактори росту — со-матомедини. Соматомедини спричиняють такі ефекти:
а) збільшують поглинання сульфатів клітинами сполучної тканини та їх включення в хондроїтинсульфат. Це викликає посилений ріст хрящів;
б) збільшують кількість мітозів і стимулюють клітинний поділ;
в) є гуморальними факторами, що беруть участь у розвитку гіпертрофії різних органів.
Метаболічна активність. СТГ має цілий ряд відносно швидких метаболічних ефектів, не пов'язаних з утворенням соматомединів. До них відносять:
а) вплив на вуглеводний обмін. СТГ, стимулюючи а-клітини острівців підшлункової залози, викликає збільшення продукції глюкагону. При тривалій дії великих доз СТГ розвивається інсулінорезистентність (див. розд. 20). Усе це в кінцевому підсумку призводить до гіперглікемії;
б) вплив на жировий обмін. СТГ, активуючи ліполіз у жировій тканині, збільшує вміст вільних жирових кислот у крові, сприяє розвитку жирової інфільтрації печінки, викликає гіперпродукцію кетонових тіл;
в) вплив на білковий обмін. СТГ має анаболічну дію. Він збільшує транспорт амінокислот у клітини і активує біосинтез білків.
2. Адренокортикотропний гормон (АКТГ, кортикотропін). Утворюється в базо-фільних клітинах аденогіпофіза. Має гландотропну і негландотропну дію. Гландотропна дія. АКТГ, впливаючи на кору надниркових залоз (в основному на пучкову зону), активує синтез і секрецію кортикостероїдних гормонів, головним чином глюкокортикоїдів.
Негландотропні ефекти:
а) посилення пігментації шкіри (відтворює дію меланоцитстимулятивного гормону);
б) мобілізація жиру з жирових депо (відтворює ефекти Р-ліпотропіну).
3. Тиреотропний гормон (ТТГ). Утворюється в базофільних клітинах передньої частки гіпофіза. Діє на щитоподібну залозу, стимулюючи утворення і вивільнення тиреоїдних гормонів.
4. Фолікулостимулятивний гормон (ФСГ). Продукується базофільними клітинами аденогіпофіза. Викликає ріст фолікулів у яєчниках і утворення естрогенів.
5. Лютеїнізуючий гормон (ЛГ). Утворюється в тих же клітинах, що і ФСГ. Разом з ним складає групу гонадотропних гормонів. Стимулює утворення жовтого тіла в яєчниках і синтез прогестинів.
6. Пролактин. Гормон еозинофільних клітин аденогіпофіза. Стимулює ріст молочних залоз і секрецію молока.
7. Меланоцитстимулюючий гормон (МСГ). Утворюється залозистими клітинами проміжної (середньої) частки гіпофіза. Стимулює утворення пігменту (меланіну) у пігментних клітинах сполучної тканини.
33.20. Що таке гіпопітуїтаризм? Які існують його форми? Чим він виявляє себе?
Гіпопітуїтаризмом називають гіпофункцію аденогіпофіза.
Розрізняють пангіпопітуїтаризм і парціальний гіпопітуїтаризм.
Пангіпопітуїтаризм — це зменшення утворення всіх гормонів аденогіпофіза. В експерименті моделюють видаленням гіпофіза (гіпофізектомією). Відомі такі клінічні форми пангіпопітуїтаризму:
1) гіпофізарна кахексія Симондса;
2) післяпологовий некроз гіпофіза — синдром Шеєгана;
3) хромофобні аденоми гіпофіза, тобто пухлини, що ростуть із хромофобних клітин. При цьому пухлина здавлює і ушкоджує залозисті клітини аденогіпофіза.
Клінічні прояви пангіпопітуїтаризму пов'язані з дефіцитом гормонів аденогіпофіза і порушенням діяльності периферичних ендокринних залоз (щитоподібної залози, кори надниркових залоз, статевих залоз). Перші симптоми ураження аденогіпофіза з'являються при ушкодженні 70-75 % тканини залози, а для розвитку повної картини пангіпопітуїтаризму необхідне руйнування 90-95 % аденогіпофіза. Синдроми, що розвиваються при тотальному порушенні функцій аденогіпофіза, представлено в таблиці:
Дефіцит гормону аденогіпофіза |
Синдроми, що розвиваються |
стг |
Відставання у рості (у дітей), раннє старіння, схильність до гіпоглікемії |
ФСГ і ЛГ |
Вторинний гіпогонадизм |
ТТГ |
Вторинний гіпотиреоз |
АКТГ |
Вторинний гіпокортицизм |
Пролактин |
Порушення лактації після пологів |
МСГ |
Депігментація |
Парціальний гіпопітуїтаризм — це порушення утворення не всіх, а окремих гормонів аденогіпофіза. Описано такі варіанти парціального гіпопітуїтаризму:
1) гіпофізарний нанізм (карликовість) — дефіцит СТГ;
2) вторинний гіпогонадизм — дефіцит ФСГ і ЛГ;
3) вторинний гіпотиреоз — дефіцит ТТГ;
4) вторинний гіпокортщизм — дефіцит АКТГ
33.21. Що таке гіперпітуїтаризм? Чим він може бути обумовлений?
Гіперпітуїтаризм - це гіперфункція аденогіпофіза. Основною причиною його розвитку є доброякісні пухлини - аденоми ендокринних клітин. Розрізняють дві групи аденом.
I. Еозинофільні аденоми. Розвиваються з ацидофільних клітин аденогіпофіза, що утворюють СТГ. Клінічно гіперфункція СТГ виявляє себе гігантизмом (якщо аденома розвивається у дітей і молодих людей до закриття епіфізарних хрящів) або акромегалією (у дорослих).
Для гігантизму характерне пропорційне збільшення всіх складових частин тіла. Акромегалія виявляється посиленим ростом акральних (кінцевих) ділянок рук, ніг, підборіддя, носа, язика, печінки.
Крім того, розвиваються ознаки підвищеної метаболічної активності СТГ — гіперглікемія, інсулінорезистентність, аж до розвитку метагіпофізарного цукрово-го діабету, жирова інфільтрація печінки.
II. Базофільні аденоми. Ростуть із базофільних клітин аденогіпофіза, найчастіше з тих, що продукують АКТГ. При цьому розвивається хвороба Іценка-Кушинга. Вона характеризується:
а) вторинним гіперкортицизмом (див. запит. 33.40);
б) посиленою пігментацією шкіри (негландотропна дія АКТГ).
Досить рідко бувають пухлини, що продукують інші гормони аденогіпофіза: ТТГ, гонадотропні гормони, пролактин, МСГ.
33.22. Які гормони виділяються при активації гіпоталамо-нейрогіпофізарної системи? Які біологічні ефекти вони мають?
Основним структурним елементом гіпоталамо-нейрогіпофізарної системи є су-праоптичне і паравентрикулярні ядра гіпоталамуса, які у відповідь на надходження інформації від рецепторів, що контролюють показники гомеостазу (осмотичний тиск, об'єм циркулюючої крові, артеріальний тиск), і від вищих нервових центрів продукують вазопресин і окситоцин. Ці гормони по аксонах нейроендокринних клітин спускаються в нейрогіпофіз, звідки надходять у.кров.
Вазопресин (антидіуретичний гормон) виявляє такі впливи:
1) діючи на дистальні звивисті канальці і збірні трубки нирок, посилює реабсорбцію води (антидіуретичний гормон);
2) викликає скорочення гладких м'язів кровоносних судин;
3) посилює глікогеноліз і глюконеогенез у печінці;
4) сприяє консолідації слідів пам'яті і мобілізації інформації, що зберігається (гормон пам'яті);
5) є ендогенним анальгетиком (пригнічує біль).
Окситоцин виявляє такі функціональні ефекти:
1) стимулює виділення молока (лактацію), викликаючи скорочення міоепітеліальних клітин дрібних проток молочних залоз;
2) ініціює й посилює скорочення вагітної матки;
3) погіршує запам'ятовування і мобілізацію інформації (амнестичний гормон).
33.23. Які розлади в організмі виникають при порушенні функції гіпоталамо-нейрогіпофізарної системи ?
1. Синдром надмірної секреції вазопресину. Виникає при пухлинах різних тканин, що утворюють вазопресин (ектопічна продукція), а також при розладах регуляції ендокринної функції гіпоталамуса. Основним його проявом є гіперволемія, що призводить до розвитку стійкої артеріальної гіпертензії.
2. Нецукровий діабет. Розрізняють два його патогенетичних варіанти: центральний (нейрогенний), при якому утворюється мало вазопресину, і нефрогений, в основі якого - нечутливість епітеліальних клітин дистальних відділів нефронів і збірних трубок до дії вазопресину (відсутність або мало У2-рецепторів).
У патогенезі основних проявів нецукрового діабету провідну роль відіграє зменшення факультативноїреабсорбції"води в нирках. Це призводить до поліурії^(добовий діурез зростає до 25 л — сечовиснаження) і зневоднення. Останнє зумовлює спрагу (полідипсію). У декомпенсованому стані зменшується об'єм циркулюючої крові (гіповолемія) і падає артеріальний тиск, розвивається гіпоксія.
3. Зменшення продукції окситоцину. Виявляє себе порушеннями лактації, слабкістю пологової діяльності.
Синдромів, обумовлених збільшенням секреції окситоцину, не описано.
Недостатність кори наднирників, гостра і хронічна: причини і механізми розвитку, патогенез основних клінічних проявів. Синдром Уотерхауса Фридерикса.
33.24. Які гормони утворюються в корі надниркових залоз?
Кора надниркових залоз гістологічно складається з трьох зон, кожна з яких синтезує свою групу гормонів.
У клубочковій (зовнішній) зоні утворюються мінеролокортикоїди - гормони, що регулюють в основному водно-сольовий обмін. Основним представником цієї групи є альдостерон, інтенсивність секреції якого становить 0,125 мг/добу.
Пучкова (середня) зона є джерелом глюкокортикоїдів — гормонів, що впливають на обмін речовин в організмі. У людини основний глюкокортикоїд — кортизол. Менше значення мають кортизон і кортикостерон. Інтенсивність секреції глюкокортикоїдів становить 20-25 мг/добу.
Клітини сітчастої (внутрішньої) зони синтезують чоловічі статеві гормони -андрогени.
33.30. Які існують порушення функції кори надниркових залоз?
Найчастіше бувають такі порушення:
1) гіпофункція кори надниркових залоз — недостатність цієї залози;
2) гіперфункція клубочкової зони — гіперальдостеронізм;
3) гіперфункція пучкової зони — синдром Іценка-Кушинга;
4) дисфункція кори надниркових залоз - адреногенітальний синдром.
33.31. Як класифікують недостатність кори надниркових залоз? Що може бути причиною гіпофункції цієї ендокринної залози?
I. За етіологією розрізняють первинну і вторинну недостатність кори надниркових залоз. Первинна виникає як результат власне ураження надниркових залоз, вторинна пов'язана або з ураженнями гіпоталамуса (дефіцит кортиколіберину), або з гіпофункцією аденогіпофіза (дефіцит АКТГ).
II. Залежно від характеру порушень виділяють тотальну (порушено утворення всіх гормонів) і парціальну (як правило, спадково обумовлені дефекти синтезу окремих гормонів) недостатність кори надниркових залоз.
III. За клінічним перебігом недостатність кори надниркових залоз може бути гострою і хронічною.
Прикладами гострої недостатності є:
а) стан після адреналектомії (видалення надниркових залоз);
б) крововиливи в надниркові залози, що виникають при сепсисі, особливо при менінгококовій інфекції (синдром Уотерхауза—Фредріксена);
в) синдром відміни глюкокортикоїдних препаратів.
Хронічна недостатність кори надниркових залоз відома під назвою хвороби Ад-дісона (бронзовоїхвороби). Найчастіші її причини:
а) туберкульозне руйнування надниркових залоз;
б) аутоімунне їх ушкодження.
33.32. Назвіть основні прояви недостатності кори надниркових залоз.
I. Прояви, пов'язані з випадінням мшералокортикоХдних функцій кори надниркових залоз:
1) зневоднення (дегідратація). Розвивається внаслідок втрати іонів натрію (зменшується реабсорбція) з наступною втратою води (поліурія);
2) артеріальна гіпотензія. Обумовлена зменшенням об'єму циркулюючої крові (результат зневоднення);
3) гемоконцентрація (згущення крові). Пов'язана із втратою рідини. Призводить до розладів мікроциркуляції й гіпоксії;
4) зменшення ниркового кровообігу (обумовлено падінням артеріального тиску) з порушенням клубочкової фільтрації і розвитком інтоксикації (азотемії);
5) гіперкаліємія. Обумовлена зменшенням канальцевої секреції іонів К+ і виходом їх з ушкоджених клітин. Викликає порушення функції збудливих тканин (див. розд. 23);
6) дистальний канальцевий ацидоз. Пов'язаний з порушенням ацидогенезу в дистальних звивистих канальцях нефронів;
7) шлунково-кишкові порушення (нуцота, блювота, проноси). У їхньому розвитку певне значення мають втрата натрію (осмотична діарея) та інтоксикація. Зазначені порушення мінералокортикоїдної функції кори надниркових залоз без відповідної корекції призводять до смерті.
II. Прояви, обумовлені порушенням глюкокортикоїдної функції кори надниркових залоз. Пов'язані з випадінням низькодозових ефектів кортизолу, а отже, усуваються введенням замісних доз глюкокортикоїдів. До таких проявів відносять:
1) гіпоглікемію, що виникає при голодуванні (непереносність голодування);
2) артеріальну гіпотензію (зменшення пермісивної дії стосовно катехоламінів);
3) зменшення реакції о/сирової тканини на звичайні ліполітичні стимули;
4) зменшення опірності організму до дії різних патогенних факторів — порушення механізмів неспецифічної резистентності (стресу);
5) зменшення здатності виводити воду при водному навантаженні (водне отруєння);
6) м 'язова слабкість і швидка стомлюваність;
7) емоційні розлади (депресія);
8) затримка росту і розвитку дітей;
9) сенсорні порушення — втрата здатності розрізняти окремі відтінки смакових, нюхових, слухових відчуттів;
10) дистрес-синдром у новонароджених (гіаліновий мембраноз). Обумовлений порушенням утворення сурфактантів у легенях, унаслідок чого легені не розправляються при народженні дитини.
ПІ. Прояви, пов'язані з впливом інших гормонів. До них, зокрема, відносять деякі ознаки, обумовлені негландотропною дією АКТГ. Зменшення концентрації глюкокортикоїдів у крові спричиняється до активації певних нейроендокринних структур гіпоталамуса і базофільних клітин аденогіпофіза. Це, у свою чергу, викликає гіперпродукцію АКТГ, який, діючи на пігментні клітини сполучної тканини, стимулює утворення меланіну і обумовлює пігментацію шкіри (звідси назва "бронзова хвороба").
Гіперфункція кори наднирників. Синдром Іценка-Кушинга. Первинний та вторинний гіперальдостеронізм. Синдром вродженої гіперплазії надниркових залоз (адреногенітальний синдром). Причини, механізми, клінічні прояви.
33.33. Що таке пперальдостєронізм? Які існують Його види? Чим він виявляється?
Пперальдостєронізм — це патологічний стан, що виникає в результаті гіперфункції клубочкової зони кори надниркових залоз, яка продукує мінералокортикоїди.
Розрізняють первинний і вторинний гіперальдостєронізм.
Первинний пперальдостєронізм (синдром Конна) виникає в результаті аденоми клубочкової зони, що утворює великі кількості альдостерону. Основні прояви цього захворювання:
1) артеріальна гіпертензія (низькоренінова). Пов'язана зі збільшенням вмісту натрію у крові і стінках кровоносних судин, унаслідок чого підвищується чутливість їхніх гладких м'язів до дії пресорних факторів, зокрема катехоламінів;
2) гіпокаліємія (результат посиленої секреції іонів К+у канальцях нирок). Вона веде до порушень діяльності збудливих органів і тканин (порушення роботи серця, міастенія, парези);
3) негазовий алкалоз. Пов'язаний з посиленням ацидогенезу в дистальних звивистих канальцях нефронів;
4) поліурія. Виникає як наслідок втрати чутливості епітелію ниркових канальців до дії вазопресину (антидіуретичного гормону). Цим, зокрема, пояснюють той факт, що при первинному гіперальдостеронізмі об'єм циркулюючої крові не зростає і набряки не розвиваються.
Вторинний гіперальдостеронізм є наслідком активації ренін-ангіотензинної системи (див. розд. 23), у процесі якої утворюються ангіотензини П і III, що діють на кору надниркових залоз. Цей стан виявляється:
а) артеріальною гіпертензією (високореніновою);
б) набряками (гіперволемічними);
в) гіпокаліємією;
г) негазовим алкалозом.
33.34. Які існують клінічні форми гіперфункції пучкової зони кори надниркових залоз? Чим вони виявляються?
Існує дві клінічні форми гіперфункції пучкової зони кори надниркових залоз. 1. Хвороба Іценка-Кушинга - базофільна аденома передньої частки гіпофіза.
2. Синдром Іценка-Кушинга:
а) пухлинний - аденома пучкової зони кори надниркових залоз;
б) ектопічна продукція АКТГ деякими злоякісними пухлинами (наприклад, рак легень);
в) ятрогенний — введення глюкокортикоїдів в організм з лікувальною метою. Гіперфункція пучкової зони кори надниркових залоз виявляє себе утворенням і
секрецією великих кількостей глюкокортикоїдів, високодозові ефекти яких і визначають клінічну картину (див. запит. 33.28). Для цього стану характерні:
1) артеріальна гіпертензія;
2) гіперглікемія — метастероїдшй іскровий діабет;
3) ожиріння;
4) інфекційні захворювання з мінімальними ознаками запалення або без них;
5) шлункова гіперсекреція і утворення виразок у шлунку і дванадцятипалій кишці;
6) остеопороз;
7) м'язова слабкість;
8) уповільнене загоєння ран.
33.35. Що таке адреногенітальний синдром? Які існують його варіанти?
Адреногенітальний синдром є відображенням дисфункції кори надниркових залоз. Він виникає в результаті спадково обумовленої блокади синтезу кортизолу і
посиленого утворення андрогенів із загальних проміжних продуктів (рис. 154).
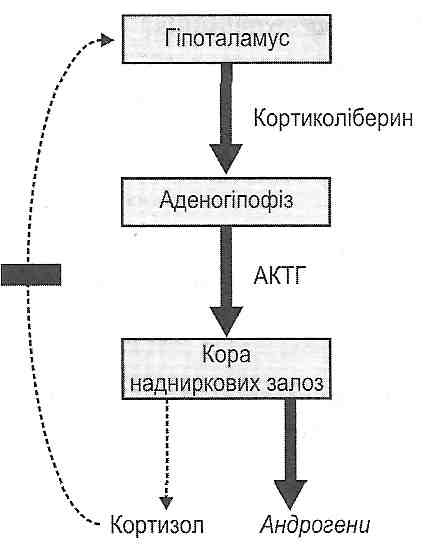
Рис. 154. Схема розвитку адреногенітального синдрому
Залежно від рівня блокади синтезу кортизолу розрізняють три варіанти адреногенітального синдрому.
I. Порушення ранніх етапів синтезу— дефіцит глюкокортикоїдів, мінералокортикоїдів і гіперпродукція андрогенів. Прояви: ознаки недостатності глюко- і мінералокортикоїд-ної функцій кори надниркових залоз, ознаки раннього статевого дозрівання в осіб чоловічої статі, вірилізація у жінок (поява чоловічих статевих ознак).
II. Порушення проміжних етапів- дефіцит глюкокортикоїдів, надлишок андрогенів, утворення мінералокортикоїдів не порушено (класичний адреногенітальний синдром). Прояви ті ж самі, що і в першому випадку, тільки без ознак недостатності мінералокор-тикоїдної функції.
III. Порушення на кінцевих етапах синтезу кортизолу — дефіцит глюкокортикоїдів, гіперпродукція андрогенів і мінералокортикоїдів. До проявів класичного адреногенітального синдрому додаються ознаки гіперальдостеронізму.
Гіпотиреоз: причини і механізми розвитку, патогенез основних клінічних проявів. Особливості гіпотиреозу у дітей.
33.44. Назвіть гормони щитоподібної залози. Як здійснюється регуляція їх утворення і секреції?
У щитоподібній залозі утворюються тиреоїдні гормони - тироксин (Т4) і три-йодтиронін (Т3). Крім того, С-клітинами (парафолікулярними) синтезується кольци-тонін, який бере участь у регуляції фосфорно-кальцієвого обміну (див. розд. 24).
Вважають, що тироксин є прогормоном трийодтироніну. На думку про це наводять такі факти:
а) Т3 в 5 разів активніший, ніж Т4;
б) ефекти Т4 розвиваються після більш тривалого латентного періоду, якщо порівнювати зі змінами, що їх викликає Т ;
в) Т4 може перетворюватися в Т3 у периферичних тканинах завдяки процесам дейо-дування.
Регуляція утворення і секреції тиреоїдних гормонів здійснюється системою гі-поталамус-аденогіпофІз за схемою: гіпоталамус → тиреоліберин → аденогіпофіз → тиреотропний гормон (ТТГ) → щитоподібна залоза. ТТГ, діючи на щитоподібну залозу, викликає такі ефекти:
а) посилює захоплення і введення йоду в органічні сполуки;
б) посилює протеоліз депонованого тиреоглобуліну;
в) посилює секрецію Т3 і Т4;
г) у разі тривалої дії викликає гіпертрофію й гіперплазію щитоподібної залози.
33.46. Назвіть основні причини гіпотиреозу.
В основі розвитку гіпофункції щитоподібної залози - гіпотиреозу - можуть лежати такі причини.
I. Центральні порушення: зменшення утворення і секреціїтиреоліберину і тире-отропного гормону (ТТГ) у зв'язку з розладами діяльності гіпоталамуса і адено-гіпофіза (вторинний гіпотиреоз).
II. Залозисті порушення, що призводять до розвитку первинного гіпотиреозу:
а) руйнування тканини залози, наприклад, радіоактивним йодом;
б) дефіцит йоду в питній воді та їжі — ендемічний зоб;
в) аутоімунне ушкодження клітин залози — аутоімунний тиреоїдит Хашимото;
г) уроджені порушення - гіпо- і аплазія щитоподібної залози, ензимопатії.
III. Периферичні порушення:
а) нечутливість периферичних клітин до дії тиреоїдних гормонів;
б) підвищене зв'язування тиреоїдних гормонів білками плазми крові;
в) посилений їх метаболізм у печінці.
33.47. Який патогенез основних проявів гіпотиреозу?
У розвитку проявів гіпотиреозу мають значення такі механізми.
I. Порушення росту і диференціювання тканий. При цьому важливу роль віді-
грають випадіння низькодозових (анаболічних) ефектів тиреоїдних гормонів і
зменшення секреції СТГ.
Оскільки тиреоїдні гормони потрібні для нормального процесу енхондральної
осифікації на межі діафіза й епіфіза, то в умовах гіпотиреозу порушується ріст
кісток у довжину. При цьому періостальний ріст кісток зберігається, у зв'язку з
чим вони стають товстими. Розвивається комплекс змін скелета — гіпотиреоїдна
карликовість.
Поряд з цим затримується і розумовий розвиток — поступово виникає кретинізм.
II. Зменшення теплоутворювальної дії тиреоїдних гормонів, що виявляється:
а) зменшенням основного обміну (падіння на 20-40 %). Воно обумовлено зниженням інтенсивності біологічного окиснення в мітохондріях і функціональної активності збудливих тканин;
б) зменшенням теплопродукції, у зв'язку з чим падає температура тіла;
в) поганою адаптацією до холоду при збереженні адаптації до високої температури;
г) гіпофагією - малим споживанням енергетичних ресурсів.
ІІІ. Зменшення функціональної активності збудливих тканий. Пов'язане з падінням активності Na-K-АТФ-аз і змінами процесів активного транспорту іонів. З другого боку, має значення зменшення чутливості тканин до катехоламінів, що обумовлено зменшенням кількості р-адренорецепторів на клітинах. Функціональні зміни збудливих органів і тканин виявляються:
а) порушеннями діяльності центральної нервової системи — уповільненням розумової діяльності, млявістю, загальмованістю, сонливістю і т. п.;
б) зменшенням функціональної активності скелетних м'язів — слабкістю, зменшенням тонусу, швидкою стомлюваністю;
в) порушеннями діяльності серцево-судинної системи - брадикардією, зменшенням хвилинного об'єму серця, падінням артеріального тиску;
г) зменшенням скорочувальної функції гладких м'язів кишок - закрепами;
ґ) порушеннями процесів всмоктування і екскреції. Зменшення всмоктування глюкози в кишках призводить до гіпоглікемії, а порушення екскреції холес-теролу в складі жовчі — до гіперхолестеролемії і атеросклерозу.
IV. Порушення з нез'ясованими механізмами розвитку. До них відносять слизовий набряк —мікседему. Характеризується збільшенням у тканинах кількості глі-козаміногліканів, що зв'язують воду; потовщенням шкіри, одутлим обличчям. Існує гіпотеза, згідно з якою мікседема є наслідком дії на сполучну тканину тире-отропного гормону (ТТГ), кількість якого при власне залозистій і периферичній формах гіпотиреозу істотно зростає.
Гіпертиреоз: причини і механізми розвитку, патогенез основних клінічних проявів.
33.48, Назвіть основні причини гіпертиреозу.
В основі розвитку гіпертиреозу можуть бути такі причини.
I. Центральні порушення - збільшення секреції тиреоліберину і тиреотропного гормону (ТТГ) при гіперфункції гіпоталамуса і аденогіпофіза (вторинний гіпертиреоз).
II. Власне залозисті порушення (первинний гіпертиреоз). Найпоширенішими клінічними формами первинного гіпертиреозу є дифузний токсичний зоб (хвороба Гревса, базедова хвороба) та аденоми щитоподібної залози.
Вважають, що дифузний токсичний зоб є аутоімунним захворюванням, у виникненні якого мають значення ТТГ-міметичні антитіла, тобто антитіла, що імітують дію ТТГ при взаємодії з поверхневими антигенами клітин щитоподібної залози (V тип алергічних реакцій за Кумбсом і Джеллом). На роль таких антитіл претендують LATS (long acting thyroid stimulator) i TSI (thyroid stimulating immunoglobuline), здатні взаємодіяти із ТТГ-рецепторами. Оскільки ці антитіла не мають місць зв'язування комплементу, то клітини щитоподібної залози не ушкоджуються, а активно функціонують.
III. Периферичні порушення:
а) збільшення чутливості клітин до дії Т3 і Т4;
б) зменшення зв'язування тиреоїдних гормонів транспортними білками;
в) уповільнення метаболізму тиреоїдних гормонів у печінці при її недостатності.
33.49. Який патогенез основних проявів гіпертиреозу?
У патогенезі проявів гіпертиреозу мають значення такі механізми. І. Антианаболічні ефекти. Є високодозовими ефектами тиреоїдних гормонів. До них відносять:
а) затримку росту;
б) атрофію м'язів і слабкість;
в) схуднення;
г) негативний азотистий баланс.
II. Посилення теплоутворювальної дії тиреоїдних гормонів. Воно виявляється:
а) збільшенням основного обміну;
б) збільшенням теплоутворення й підвищенням температури тіла;
в) збереженою адаптацією до холоду і поганою — до високої температури;
г) гіперфагією - підвищеним споживанням енергетичних ресурсів.
III. Збільшення функціональної активності збудливих тканин. Пов'язане з підвищенням активності Na-K-насосів клітинних мембран і збільшенням чутливості клітин до катехоламінів. Цим, зокрема, обумовлені такі прояви гіпертиреозу:
а) порушення діяльності центральної нервової системи — прискорення психічних процесів, занепокоєння, збудження, безсоння;
б) постійна спонтанна скорочувальна активність волокон скелетних м'язів -фібрилярні посмикування, тремор. З цим пов'язана м'язова слабкість, стомлюваність;
в) зміни діяльності серцево-судинної системи - тахікардія, збільшення хвилинного об'єму серця, артеріального тиску;
г) підвищення скорочувальної активності гладких м'язів кишок — проноси;
ґ) збільшення інтенсивності процесів усмоктування і екскреції. З цим, зокрема, пов'язана гіперглікемія і гіпохолестеролемія.
IV. Катехоламінові ефекти. Обумовлені підвищенням чутливості клітин до дії катехоламінів у зв'язку зі збільшенням на клітинній поверхні кількості )3-адреноре-цепторів.
У клініці гіпертиреозу велике значення мають функціональні ефекти катехоламінів, зокрема, їхній вплив на серцево-судинну систему (див. запит. 33.36), і метаболічні зміни. З останніми пов'язані такі порушення:
а) посилення глікогенолізу в печінці → гіперглікемія → гіперфункція β-клітин острівців підшлункової залози з наступним їх виснаженням → тиреогдний цукровий діабет',
б) посилення ліполізу в жировій тканині → гіперліпацидемія → збільшення кетогенезу в печінці → метаболічний ацидоз;
в) активація не пов'язаного з фосфоруванням окиснення в бурій жировій тка^ нині → збільшення теплопродукції —> підвищення температури тіла і основного обміну.
V. Порушення з нез'ясованими механізмами розвитку- орбітопатія і двосто-
ронній екзофтальм (витрішкуватість). В основі їхнього розвитку — набряк і лім-фоїдна інфільтрація м'язів очного яблука і ретробульбарної тканини. Ці зміни не залежать від концентрації тиреоїдних гормонів у плазмі крові і від рівня ТТГ-мі-метичних антитіл (LATS, TSI). Вважали, що в умовах гіпертиреозу виділяється особливий екзофтальмічний фактор, однак його дотепер не виявлено.
Зоб: види, етіологія, патогенез; порушення функціонального стану щитоподібної залози. Особливості у дітей.
33.50. Що таке зоб? Які його види виділяють? Який патогенез ендемічного зоба?
Зобом називають видиме збільшення щитоподібної залози. Виділяють три види зоба.
1. Дифузний токсичний зоб- гіпертиреоїдний (хвороба Гревса, базедова хвороба). Характеризується ознаками гіперфункції щитоподібної залози.
2. Спорадичний зоб — еутиреоїдний. Збільшення маси щитоподібної залози не супроводжується вираженими змінами її функціональної активності.
3. Ендемічний зоб - гіпотиреоїдний. Виявляє себе клінікою гіпофункції щитоподібної залози.
Причиною ендемічного зоба є недостатній вміст йоду в питній воді і продуктах харчування, що пов'язано з особливостями ґрунту і підземних вод у певних регіонах і місцевостях земної кулі. Дефіцит йоду призводить до порушення утворення тире-оїдних гормонів, вміст яких у крові зменшується. Це викликає посилення продукції тиреоліберину і тиреотропного гормону (ТТГ). Останній, впливаючи на тканину щитоподібної залози, стимулює процеси гіпертрофії і гіперплазії — розвивається зоб. Оскільки збільшення залози не усуває дефіцит тиреоїдних гормонів (причину — недостатність йоду - не ліквідовано), то підвищена продукція ТТГ зберігається і триває його дія на тканину залози: зоб прогресує.
Гіпо- та гіперфункція паращитоподібних залоз: етіологія, патогенез, типові порушення в організмі.
33.51. Як здійснюється регуляція утворення і секреції паратирину прищитоподібними залозами? Назвіть основні біологічні ефекти
паратгормону.
Утворення і секреція паратирину (паратгормону) регулюється вмістом іонів кальцію в плазмі крові. Секреція цього гормону зростає при зменшенні концентрації іонів Са2+ у плазмі, і навпаки, зменшується при збільшенні вмісту цих іонів. Крім того, вивільнення паратирину в кров пригнічує 24,25-(ОН)2 вітамін D, що утворюється в нирках (див. розд. 24).
Біологічні ефекти паратирину (докладно див. розд. 24):
1. Дія на кісткову тканину - активація функції остеокластів.
2. Дія на нирки — пригнічення реабсорбції фосфату.
3. Активація перетворення в нирках вітаміну D у його гормональну форму— 1,25-(ОН)2 вітамін D.
Наслідком усіх зазначених ефектів є збільшення концентрації іонів кальцію в плазмі крові.
33.52. Які причини і чим виявляє себе гіпопаратиреоз?
Причини гіпопаратиреозу -гіпофункції прищитоподібних залоз:
1) випадкове ушкодження або видалення прищитоподібних залоз при операціях на щитоподібній залозі;
2) ушкодження прищитоподібних залоз при лікуванні радіоактивним йодом хвороб щитоподібної залози;
3) аутоімунні ушкодження прищитоподібних залоз;
4) уроджене недорозвинення прищитоподібних залоз;
5) нечутливість клітин-мішеней до дії паратирину — псевдогіпопаратиреоз.
Основним проявом гіпопаратиреозу є гіпокальціємія. Вона обумовлює розвиток паратиреопривної тетанії, що виявляється різким підвищенням нервово-м'язової збудливості, множинними фібрилярними скороченнями м'язів усього тіла. Потім
виникають напади клонічних судом, що переходять у тонічні. Судомні скорочення можуть поширюватися і на внутрішні органи (пілороспазм, ларингоспазм). Під час одного з таких нападів настає смерть.
При хронічному гіпопаратиреозі у тварин розвивається клінічна картина парати-реопривної кахексії. Вона характеризується схудненням, анорексією, підвищеною нервово-м'язовою збудливістю, диспепсією й різноманітними трофічними порушеннями.
33.53. Які причини і чим виявляє себе гіперпаратиреоз?
Причини гіперпаратиреозу — гіперфункції прищитоподібних залоз:
1) пухлина — аденома прищитоподібної залози;
2) гіперфункція прищитоподібних залоз, обумовлена зменшенням чутливості їхніх ендокринних клітин до іонів кальцію, — порушення регуляції за принципом негативного зворотного зв'язку.
Гіперпаратиреоз виявляється двома групами пов'язаних між собою змін.
І. Порушення кісткової тканини- генералізована фіброзна остеодистрофія. Обумовлена підвищенням активності остеокластів і пригніченням функції остеобластів. Виявляється болем у кістках і суглобах, розм'якшенням кісток, різкою деформацією скелета. Розвивається демінералізація кісткової тканини (остеомаляція), що обумовлює підвищення вмісту іонів кальцію в плазмі крові — гіпер-кальціємію.
II. Гіперкальціємія. З нею пов'язані:
а) кальцифікація м'яких тканин (нирок, судин, легень). У важких випадках розвивається ниркова недостатність;
б) утворення кальцієвих каменів у нирках;
в) порушення збудливості нервової системи і м'язів - м'язова слабкість, депресія, порушення пам'яті;
г) артеріальна гіпертензія;
ґ) посилення шлункової секреції.
Порушення функції статевих залоз: первинні та вторинні стани гіпер- і гіпогонадизму. Етіологія, патогенез, типові клінічні прояви.
33.54. Як здійснюється регуляція утворення і секреції чоловічих статевих гормонів? Назвіть їх основні біологічні ефекти.
Ендокринні клітини сім'яників (інтерстиціальні клітини, або клітини Лейдига) синтезують і секретують чоловічі статеві гормони - андрогени, серед яких основним є тестостерон.
Регуляція утворення андрогенів здійснюється за участю системи гіпоталамус-аденогіпофіз за схемою: гіпоталамус → гонадоліберин → аденогіпофіз → лютеї-нізуючий гормон → інтерстиціальні клітини сім'яників → тестостерон → клітини Сертолі сім'яників → інгібін → гальмування функціональної активності відповідних утворень гіпоталамуса і аденогіпофіза → зменшення утворення гонадоліберину і лютеїнізуючого гормону → зменшення синтезу і секреції андрогенів → активація гіпоталамуса і т. д.
Основні біологічні ефекти тестостерону: 1. Ембріональне диференціювання. Андрогени забезпечують диференціювання статевих залоз і статевих органів у період ембріонального розвитку. Утворюючись
під впливом хоріонічного гонадотропіну (у період між 6 і 16 тижнями), тестостерон активує формування чоловічих статевих органів з вольфової протоки, урогеніталь-ного синуса і горбка, одночасно пригнічуючи розвиток жіночих статевих органів. Крім того, андрогени забезпечують диференціювання структур центральної нервової системи "за чоловічим типом", визначаючи надалі чоловічий тип статевої поведінки.
2. Статеве дозрівання і розвиток вторинних статевих ознак в особин чоловічої статі. Оскільки андрогени стимулюють діяльність сальних залоз, то в пубертатному періоді шкіра стає жирною, часто відбувається інфікування сальних залоз - з'являються вугрі.
3. Регуляція сперматогенезу. Тестостерон активує діяльність міоепітеліальних клітин сім'яників (клітин Сертолі) та епітеліальних клітин проток, які у свою чергу здійснюють вплив на інтенсивність поділу статевих клітин та їхнє дозрівання. Така дія тестостерону не пов'язана з надходженням його в кров, а є місцевою.
4. Регуляція статевої поведінки. Андрогени мають стосунок до формування статевого потягу (лібідо) і здатності до копуляції. Це пов'язано з тим, що тестостерон впливає на преоптичну зону гіпоталамуса (центр статевої поведінки) і полегшує спинномозкові рефлекси, важливі для копуляції. Тимчасова недостатність андрогенів у період статевого диференціювання і розвитку мозку призводить до формування в подальшому гомосексуальної орієнтації.
5. Анаболічна дія. Тестостерон активує процеси білкового синтезу. Саме з цим пов'язаний вплив андрогенів на ріст скелета і м'язів (посилення росту кісток у довжину, збільшення м'язової маси).
33.55. Які причини й основні прояви чоловічого гіпогонадизму?
Чоловічий гіпогонадизм — це гормональна недостатність чоловічих статевих за-
лоз. Його причинами можуть бути:
ї. Центральні (дисрегуляторні) порушення-зменшення утворення гонадолі-берину в гіпоталамусі, лютеїнізуючого гормону в аденогіпофізі. Це може бути пов'язано з ураженням зазначених структур, а також з гіперфункцією епіфіза, у разі якої збільшення утворення мелатоніну супроводжується пригніченням синтезу гонадотропних гормонів.
II. Власне залозисті порушення:
а) кастрація — видалення сім'яників;
б) фіброз яєчок після деяких вірусних інфекційних захворювань, ускладнених орхітом (наприклад, після епідемічного паротиту);
в) порушення розвитку яєчок.
Після втрати яєчок розвивається комплекс порушень за назвою євнухізм. Гіпофункцію яєчок при їх збереженні позначають терміном "євнухоїдизм".
III. Периферичні порушення:
а) зменшення чутливості клїтин-мтшенеи до дії андрогенів;
б) збільшене зв'язування тестостерону з білками плазми крові;
в) посилене руйнування андрогенів у печінці.
Прояви чоловічого гіпогонадизму залежать від того, у який період онтогенезу він розвивається.
Гіпогонадизм до періоду статевого дозрівання може виявлятися:
• синдромом тестикулярної фемінізації (спадково обумовленим чоловічим псевдо-гермафродитизмом). Цей синдром обумовлений порушеннями диференціювання статевої системи за чоловічим типом в період ембріогенезу;
• слабко вираженими вторинними статевими ознаками;
• пригніченням сперматогенезу;
• відсутністю статевого потягу;
• порушенням окостеніння епіфізарних хрящів при збереженому впливі СТГ — розвивається євнухоїдний, або гіпогонадний, гігантизм;
° слабким розвитком скелетної мускулатури (відсутність анаболічних ефектів андрогенів). Гіпогонадизм після завершення статевого дозрівання характерний, зокрема, для процесу старіння. Він виявляється:
• деяким зменшенням вираженості вторинних статевих ознак;
• порушеннями сперматогенезу;
• розвитком імпотенції;
• анаболічними порушеннями — зменшенням маси скелетних м'язів і працездатності.
33.56. Які причини і основні прояви чоловічого гіпергонадизму?
Чоловічий гіпергонадизм може розвиватися в дитячому віці до періоду статевого дозрівання і у дорослих.
У дітей гіпергонадизм найчастіше пов'язаний з пухлинами і запальними процесами в гіпоталамусі, а також гіпофункцією епіфіза. Усі ці причини викликають збільшення продукції гонадотропних гормонів і, як наслідок, андрогенів. Клінічно такий гіпергонадизм виявляється передчасним статевим дозріванням.
Причиною гіперфункції статевих залоз у дорослих є добро- і злоякісні пухлини, що ростуть з інтерстиціальних клітин. При цьому виражених клінічних ознак, пов'язаних з гіперандрогенемією, немає. У випадку злоякісних новоутворень характерні відсутність кахексії, збереження м'язової маси і сили м'язів.
33.57. Які гормони відносять до жіночих статевих? Як здійснюється регуляція їх утворення і секреції?
Місцем синтезу жіночих статевих гормонів є яєчники. Тут утворюється три групи гормонів.
I. Естрогени: естрадіол, естрон, естріол. Продукуються епітеліальними клітинами фолікулів.
II. Прогестіши, серед яких основним є прогестерон. Утворюються клітинами жовтого тіла.
III. Мінорні гормони: інгібін (пригнічує утворення ФСГ в аденогіпофізі) ірелаксин (розм'якшує лобкове зрощення, тазові зв'язки, шийку матки, розслаблює гладкі м'язи матки).
У регуляції утворення і секреції жіночих статевих гормонів вирішальне значення мають система гіпоталамус-аденогіпофіз-яєчники і механізми позитивного та негативного зворотного зв'язку.
У передовуляційний період під впливом ФСГ посилюється утворення і секреція естрогенів, які за принципом позитивного зворотного зв'язку стимулюють утворення гонадоліберину в гіпоталамусі і лютеїнізуючого гормону (ЛГ) в аденогіпофізі (забезпечують "пік ЛГ"). Останній викликає підвищення секреторної активності клітин, що продукують прогестини.
У післяовуляційний період естрогени і прогестини за принципом негативного зворотного зв'язку пригнічують утворення гонадоліберину і гонадотропних гормонів аденогіпофіза, що веде в підсумку до зменшення секреції і самих жіночих статевих гормонів.
33.58. Назвіть основні біологічні ефекти жіночих статевих гормонів.
I. Статеве дозрівання і розвиток вторинних статевих ознак в особин жіночої статі. Ці ефекти обумовлені естрогенами, секреція яких починає зростати в період статевого дозрівання.
II. Забезпечення! циклічних змін в організмі жінки — підготовка статевих органів до запліднення та імплантації яйцеклітини. Це здійснюється завдяки участі жіночих статевих гормонів разом з ФСГ і ЛГ у гормональному контролі овуляції й менструального циклу.
III. Забезпечення вагітності, пологів і лактації. Так, естрогени викликають швидкий ріст м'язів матки, стимулюють ріст системи проток молочних залоз. Прогестини блокують скорочувальну активність вагітної матки, стимулюють ріст залозистого епітелію молочних залоз. Зі зменшенням продукції прогестинів плацентою пов'язують настання пологів.
IV. Екстра генітальні ефекти. Естрогени впливають на:
а) кісткову тканину, забезпечуючи умови для своєчасного окостеніння епіфі-зарних хрящів;
б) печінку, активуючи синтез цілого ряду білків (транспортних білків, факторів зсідання, ангіотензиногену, ліпопротеїдів високої густини);
в) кровоносні судини, регулюючи процеси ангіогенезу, судинний тонус.
З дією прогестинів пов'язують збільшення базальної температури тіла в післяовуляційний період.
33.59. Які причини і основні прояви жіночого гіпогонадизму?
Виділяють такі групи причин жіночого гіпогонадизму.
I. Центральні (дисрегуляторнї) порушення. Можуть бути обумовлені психогенними факторами, ураженнями гіпоталамуса (дефіцит гонадоліберину), гіпофункцією аденогіпофіза (дефіцит ФСГ і ЛГ), гіперфункцією епіфіза (зменшення утворення гонадотропних гормонів при збільшенні синтезу мелатоніну).
II. Власне залозисті порушення:
а) спадково обумовлена аплазія яєчників;
б) дегенерація яєчників (запальна, кістозна);
в) аутоімунне ушкодження жіночих статевих залоз;
г) хірургічне видалення яєчників. III. Периферичні розлади:
а) зменшення чутливості клітин-мішеней до дії жіночих статевих гормонів;
б) підвищене зв'язування їх білками плазми крові;
в) посилене руйнування жіночих статевих гормонів у печінці.
Клінічні прояви гіпофункції статевих залоз залежать від часу настання гіпо-гонадизму.
Якщо він розвивається до настання статевого дозрівання, то формується комплекс порушень під назвою "оваріальний євнухоїдизм": слабкий розвиток вторинних статевих ознак, первинна аменорея, високий зріст.
Розвиток гіпогонадизму в дітородному віці супроводжується порушеннями циклічних процесів в організмі жінки (розлади менструального циклу), безплідністю, передчасним клімаксом.
Після менопаузи розвиваються клімактеричні зміни - нестабільність судинного тонусу, остеопороз та ін.
33.60. Які причини і основні прояви жіночого гіпергонадизму?
В основі жіночого гіпергонадизму, при якому збільшується секреція жіночих статевих гормонів, можуть лежати центральні (дисрегуляторні) порушення і власне залозисті причини.
Центральні порушення, обумовлені збільшенням секреції гонадоліберину і гонадотропних гормонів, можуть виявлятися розвитком синдрому передчасного статевого дозрівання і синдрому уявної вагітності.
Для першого характерні рання поява вторинних статевих ознак, ранній початок менструацій і ймовірність завагітніти (у віці 7—8 років). Другий синдром виявляється аменореєю й усіма зовнішніми ознаками вагітності. При цьому має місце підвищена продукція пролактину клітинами аденогіпофіза.
До власне залозистих причин жіночого гіпергонадизму відносять кістозні процеси і пухлини яєчників, що продукують гормони. При цьому розвивається гіпертрофія ендометрію, порушується менструальний цикл, поновлюються маткові кровотечі в менопаузі.
Стрес. Визначення поняття, причини та механізми розвитку, стадії. Поняття про «хвороби адаптації».
33.38. Що таке стрес і загальний адаптаційний синдром?
Стрес - це стан напруження неспецифічних адаптаційних механізмів, що виникає у разі дії на організм надмірних за силою або патогенних факторів (рис. 155).
Клінічно стрес виявляє себе комплексом структурних, функціональних і біохімічних змін, що отримали назву загального адаптаційного синдрому.
Термін "стрес" уперше ввів у вжиток Г. Сельє у 1936 р.
33.39. Які морфологічні ознаки характерні для загального адаптаційного синдрому?
Дія на організм численних патогенних факторів, незалежно від їхніх властивостей і походження, дає стандартну відповідь — так звану морфологічну тріаду:
1) гіпертрофію кори надниркових залоз;
2) інволюцію тиміко-лімфоцитарної системи (атрофію вилочкової залози і лімфатичних вузлів);
3) утворення виразок і ерозій у шлунку і в кишках.
Рис. 155. Фактори, що викликають стрес
33.40. Які стадії виділяють у розвитку стресу?
I. Стадія тривоги:
а) підстадія шоку. Характеризується короткочасним зменшенням резистентності до патогенного фактора;
б) підстадія контршоку. Опірність організму спочатку поновлюється, а потім підвищується.
II. Стадія резистентності. Характеризується стійким і тривалим збільшенням опірності організму як до фактора, що викликав стрес, так і до інших патогенних агентів.
III. Стадія виснаження. Настає при дуже інтенсивній або тривалій дії патогенного фактора, а також в умовах функціональної слабкості адаптаційних механізмів. Супроводжується зменшенням резистентності організму до патогенних впливів (рис. 156). '
Рис. 156. Стадії загального адаптаційного синдрому
33.41. Які механізми беруть участь у реалізації стресу?
Численні ініціатори стресу (cmpecopu) - травма, холод, біль, емоції, кровотеча, фізичне навантаження, гіпоглікемія, інфекції та ін. — через порушення гомеостазу або передвісників такого порушення викликають збудження вищих нервових регупя-
торних центрів і пов'язане з цим вивільнення великої кількості гормонів. При цьому велике значення мають такі процеси.
I. Активація системи гіпоталамус-аденогіпофіз. Як наслідок відбувається виділення АКТГ, СТГ, ТТГ, які відповідно стимулюють секрецію глюкокортикоїдів, соматомединів, тиреоїдних гормонів.
II. Активація вегетативної нервової системи (симпатичної і парасимпатичної) супроводжується надходженням у кров катехоламінів, інсуліну, глюкагону.
III. Активація альдостерон-вазопресинової системи веде до збільшення вмісту в крові ангіотензинів, альдостерону, вазопресину (АДГ) (рис. 157).
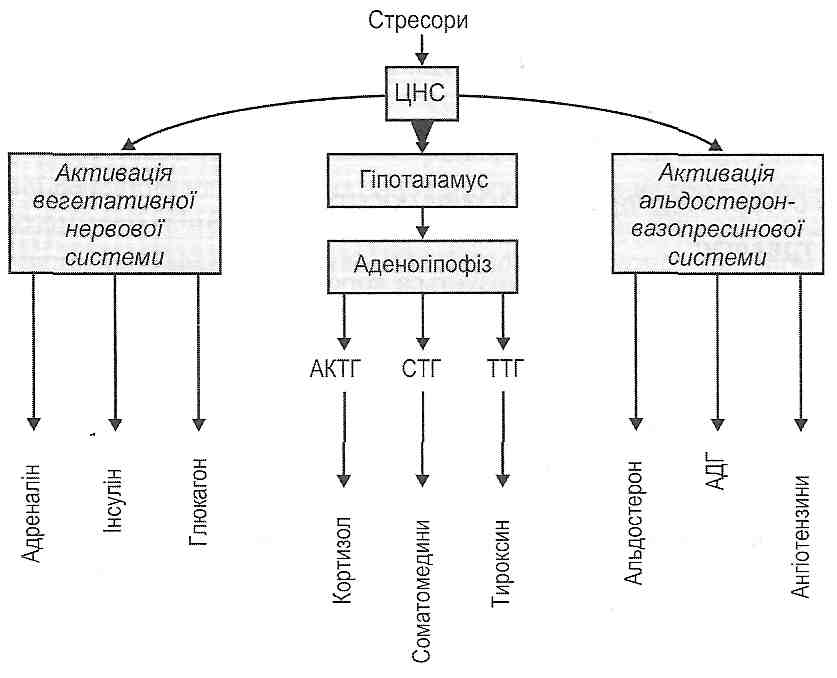
Рис. 157. У реалізації стресу беруть участь різні гормони
33.42. Яка участь різних гормонів у розвитку стресу?
Гормони, що виділяються під час стресу, визначають розвиток трьох послідовних фаз цієї реакції.
І. Гостра фаза. Гормони, що вивільнюються в цю фазу, забезпечують захист від падіння артеріального тиску і об 'ему циркулюючої крові. Це досягається збільшенням загального периферичного опору і збереженням води в організмі. Зазначені реакції пов'язані з посиленим надходженням кров катехоламінів, глюкокортикоїдів, ангіотензину II, альдостерону, вазопресину. її. Підгостра фаза. Характеризується мобілізацією ресурсів для енергетичного й пластичного забезпечення систем, що здійснюють адаптацію. Цьому слугує перерозподіл зазначених ресурсів між активно функціонуючими органами (серце, головний мозок) і структурами, що перебувають у відносному спокої (скелетні
м'язи, травний канал, лімфоїдна і жирова тканини). Метаболічні зміни в цю фазу обумовлені збільшенням секреції катехоламінів, глюкокортикоїдів, глюкагону і зменшенням виділення інсуліну. Наведені вище контрінсулярні гормони, посилюючи глікогеноліз, глюконеогенез, ліполіз і протеоліз, викликають збільшення вмісту в крові глюкози, амінокислот, вільних жирових кислот. III. Фаза довгострокової адаптації. Характеризується структурними змінами (гіпертрофією, гіперплазією) органів і тканин, що забезпечують адаптацію і перебувають у стані гіперфункції. У її реалізації беруть участь інсулін, СТГ, соматомедини, тиреоїдні гормони, фактори росту - істинні та тканинні гормони, що активують анаболічні процеси і формують так званий структурний слід адаптації.
33.43. Що таке "хвороби адаптації"? Які захворювання до них відносять ?
Хвороби адаптації- це захворювання, у розвитку яких провідна роль належить надмірному стресу і так званим стресорним механізмам ушкодження.
При великій інтенсивності і тривалості стрес із механізму адаптації може перетворитися у механізм патогенезу.
До хвороб адаптації відносять:
а) психосоматичні захворювання (ішемічну хворобу серця, гіпертонічну хворобу, виразкову хворобу шлунку і дванадцятипалої кишки);
б) хвороби обміну речовин (цукровий діабет);
в) алергічні і запальні захворювання (бронхіальну астму, ревматизм).
Загальна характеристика патології нервової системи, принципи класифікації порушень її діяльності. Особливості розвитку типових патологічних процесів у нервовій системі. Роль змін гематоенцефалічного бар'єру в патогенезі порушень діяльності ЦНС.
34.1. Як класифікують порушення діяльності нервової системи?
І. За анатомічним принципом виділяють:
1) порушення периферичної нервової системи;
2) порушення центральної нервової системи, у тому числі розлади функції спинного мозку, довгастого, середнього і т. д.
її. За походженням виділяють спадково обумовлені і набуті порушення нервової системи. Набуті можуть бути первинними і вторинними.
Первинні розлади виникають унаслідок прямої дії на нервову систему патогенних факторів: фізичних (травма, радіація, термічні впливи), хімічних (токсини, отрути), біологічних (віруси, бактерії), соціальних (слово). Вторинні розлади обумовлені насамперед порушеннями гомеостазу (гіпоксія, гіпоглікемія, ацидоз і т. п.), імунними факторами (аутоалергічні реакції), розладами мозкового кровообігу.
III. Клітинний принцип передбачає такі види порушень функції нейронів:
1) порушення електрофізіологічних процесів;
2) розлади нейрохімічних (медіаторних) процесів;
3) порушення аксоплазматичного транспорту.
IV. Залежно від виду порушених функцій розрізняють такі розлади діяльності нервової системи:
1) порушення сенсорних функцій (чутливості);
2) порушення ефекторних функцій: рухової, вегетативної, трофічної;
3) порушення інтегративних функцій.
Порушення сенсорної функції нервової системи. Розлади механо-, термо-, пропріо- і ноцицепції. Порушення проведення сенсорної інформації. Прояви ушкодження таламічних центрів і сенсорних структур кори головного мозку.
34.2. Які виділяють види порушень соматовісцеральної чутливості?
Поняття соматовісцеральної чутливості охоплює чутливість шкіри (тактильну, температурну, больову), глибоку чутливість (пропріорецепцію) і больову чутливість усього тіла (ноцицепцію).
Розрізняють такі види порушень соматовісцеральної чутливості:
1) гіперестезія — підвищення чутливості;
2) гіпестезія - зменшення чутливості;
3) анестезія — відсутність чутливості.
34.3. Які провідникові шляхи забезпечують надходження інформації про різні види чутливості від периферичних рецепторів у центральну нервову систему?
1. Лемнісковий шлях (рис. 159). Проводить усі види глибокої (пропріоцептивної) чутливості, а також тактильну чутливість від спеціалізованих механорецепторів шкіри (складна тактильна чутливість). Складається з трьох нейронів.
Рис. 159. Схема лемніскового
шляху
Рис. 160. Схема антеро-латерального шляху
Перші нейрони (І) - псевдоуніполярні клітини спинномозкових гангліїв. Аксони цих клітин входять через задні корінці в спинний мозок і піднімаються догори в складі задніх канатиків, утворюючи пучки Голля і Бурдаха.
Тіла других нейронів (II) містяться в довгастому мозку (n. gracilis і n. cuneatus). їхні аксони переходять на протилежний бік, перехрещуючись з аксонами контр-латеральних нейронів, і утворюють медіальну петлю (lemniscus medialis). Тіла третіх нейронів (III) містяться в таламусі. їхні аксони йдуть у кору головного мозку, у відповідні сенсорні зони.
2. Антеро-латеральний шлях (неоспіноталамічний тракт) (рис. 160). Проводить температурну, найпростіші види тактильної (від механочутливих вільних нервових закінчень) і шкірну больову (ранній біль) чутливість.
Перші нейрони (І) є клітинами спинномозкових гангліїв. Тіла других (II) нейронів містяться в задніх рогах спинного мозку. їхні аксони посегментно переходять на протилежний бік спинного мозку, перехрещуються і у складі бічних канатиків піднімаються в таламус, де містяться тіла третіх нейронів (III), що посилають свої відростки в сенсорні зони кори головного мозку.
3. Екстралемнісковий шлях (рис. 161). Проводить больову чутливість (пізній біль, глибокий і вісцеральний біль). На відміну від двох попередніх, є багатонейронним і філогенетично більш давнім.
Тіла перших нейронів (І) містяться в спинномозкових гангліях, а других (II) - у задніх рогах спинного мозку. Аксони останніх частково переходять на другий бік, а частково з цього ж боку йдуть нагору в складі бічних канатиків, утворюючи два тракти: спіноретикулярний і палеоспіноталамічний.
У ретикулярній формації стовбура мозку, куди піднімаються аксони других нейронів, відбувається багаторазове перемикання на інші нервові клітини. Від ре-
Рис. 161. Схема екстралемніскового шляху
тикулярної формації інформація надходить в утвори лімбічної системи (емоційні компоненти реакцій), центри гіпоталамуса (вегетативні компоненти реакцій), таламус, кору головного мозку та ін.
34.4. Які механізми можуть лежати в основі порушень соматовісцеральної чутливості?
1. Порушення рецепції. При збільшенні порогу збудження рецепторів виникає гіпестезія, при зменшенні - гіперестезія.
2. Ушкодження периферичних нервів. При цьому випадають усі види чутливості в зоні іннервації даного нерва. Як правило, зона випадання чутливості менша зони іннервації, що пояснюють деяким перекриванням зон іннервації різних нервів.
3. Ушкодження задніх корінців спинного мозку. Характеризується випаданням усіх видів чутливості в зоні відповідних сегментів.
4. Ушкодження спинного мозку. При перетинанні половини спинного мозку (лівої або правої) розвивається
синдром Броун-Секара (див. запит. 34.5). Повний перетин спинного мозку веде до зникнення усіх видів чутливості по обидва боки нижче рівня перетинання.
5. Порушення функції підкіркових структур, що беруть участь у здійсненні сенсорних функцій. Найбільше значення має ураження ядер таламуса.
6. Ураження сенсорних зон кори головного мозку. Порушення нейронів постцен-тральної звивини спричиняються до розладів складної тактильної і пропріоцеп-тивної чутливості на протилежному боці тіла. Ушкодження тім'яної частки викликає розвиток комплексу порушень під назвою "аморфосинтез".
У людини (що не є шульгою) аморфосинтез виявляють після видалення кори правої півкулі великого мозку. При цьому зникає уява про просторове розташування частин тіла на протилежному боці. Людина не може надягти одяг або привести його в порядок на лівій половині, не може поголити ліву половину обличчя або зачесати волосся на лівому боці.
Якщо ж уражено тім'яну частку з лівого боку, то аморфосинтез доповнюється агнозією - нездатністю розпізнавати частини тіла, предмети, їхнє зображення і розташування в просторі.
34.5. Коли виникає синдром Броун-Секара? Чим він виявляється?
Синдром Броун-Секара розвивається після перетинання половини спинного мозку (лівої або правої). Характеризується дисоціацією розладів чутливості. Так, нижче рівня перетину з того ж боку випадають пропріоцептивна і складні види тактильної чутливості (ушкоджується лемнісковий шлях до його перехрещення), а з протилежного боку — температурна, проста тактильна й частково больова чутливість (ушкоджується антеро-латеральний шлях після перехрещення).
Біль. Принципи класифікації. Соматичний біль. Сучасні уявлення про причини і механізми розвитку болю: теорія розподілу імпульсів, теорія специфічності. Патологічний біль. Реакції організму на біль. Природні антиноцицептивні механізми.
Біль - неприємне сенсорне і емоційне відчуття, пов’язане із загрозою або самим ушкодженням тканин.
Особливості болю як виду чутливості:
1) Біль дає мало інформації про навколишній світ, проте інформує про небезпеку, яка може виникнути або уже виникла внаслідок дії ушкоджуючих факторів - захисна функція болю.
2) На відміну від інших видів чутливості до болю не розвивається адаптація. У зв’язку з цим біль може бути причиною страждань хворого.
3) Біль супроводжується складними емоційними, вегетативними і руховими реакціями.
4) Біль може бути патогенетичним механізмом розвитку генералізованих патологічних процесів, зокрема шоку.
Класифікація болей.
1) За клінічною характеристикою (суб’єктивним відчуттям) біль може бути: а) гострим і тупим, б) локалізованим і дифузійним, мати характер пощипування, поколювання, жару і т.п.
2) У залежності від тривалості больових відчуттів розрізняють біль: а) гострий - швидко проходить після припинення дії больових стимуляторів, б) хронічний – тривалий біль, який викликає страждання хворого.
3) За значенням для організму біль може бути: а) фізіологічним і б) патологічним.
Фізіологічний біль має захисне значення - сигналізує про ушкодження або його можливість, сприяє включенню визначених поведінкових реакцій, спрямованих на усунення ушкодження, обмежує функції ушкодженого органа.
Патологічний біль не несе сигнальної функції, він стає механізмом порушення життєдіяльності того чи іншого органа (у тому числі мозку), приводить до розладів функції різних органів і систем.
4) За місцем виникнення розрізняють: а) соматичний (поверхневий і глибокий) і б) вісцеральний біль.
Соматичний поверхневий біль - виникає у шкірі. Розрізняють його 2 види: а) раннійі б) пізній біль.
Ранній біль - гострий, різкий, добре локалізований біль, який виникає відразу ж після нанесення ушкодження і швидко проходить після завершення дії патогенного фактора. Характеризується: а) низьким порогом больового відчуття, б) градуальністю (ступінь болю зростає із збільшенням інтенсивності ушкодження), в) проводиться мієлінізованими волокнами групи А із швидкістю 10-25 м/с, г) проводиться пердньо-латеральним неспинно-таламічним шляхом, д) формується в сенсорних центрах кори великого мозку, е) вегетативним компонентом є порушення симпатичної нервової системи, є) є засобом попередження про ушкодження, і) має захисне значення - включає захисні рефлекси і реакції, к) є епікритичною (еволюційно більш молодою).
Пізній біль - тупий, ниючий, дифузний, виникає через певний час (0,5-1с), зникає поступово. Характеризується: а) високим порогом больового відчуття, б) відсутністю градуальності - закон "все або нічого", в) проводиться немієлінізованими волокнами групи С із швидкістю 0,5—1 м/с, г) проводиться екстралемнісковим шляхом, д) формується у таламічних сенсорних центрах, е) вегетативним компонентом є порушення парасимпатичної нервової системи, є) є засобом нагадування про ушкодження, і) відіграє патогенну роль - викликає афективні (емоційні) реакції, загальне нездужання, хворобливий стан, к) є протопатичною (еволюційно більш давньою).
Соматичний глибокий біль - виникає в глибоких тканинах. До нього відноситься: а) головний біль, б) зубний біль, в) біль у м’язах і суглобах.
Має багато спільного з пізнім соматичним поверхневим болем: а) часто тупий, б) не має чіткої локалізації, в) супроводжується афектними (загальне нездужання, хворобливий стан) і вегетативними (нудота, потовиділення, зменшення артеріального тиску) реакціями.
Вісцеральний біль - виникає у внутрішніх органах.
Для вісцерального болю х а р а к т е р н і:
1) афективні реакції (пригнічений емоційний стан, загальне нездужання, стан хвороби);
2) вегетативні реакції (нудота, потовиділення, падіння артеріального тиску);
3) рефлекторне скорочення скелетних м’язів (напруга м’язів черевної стінки, вимушена поза і ін.).
У залежності від локалізації виділяють наступні види болю:
1) Місцевий біль - локалізується в місці дії подразника, у ділянці розвитку патологічного процесу.
2) Проекційний біль - місце, на яке діє больовий подразник, не збігається із місцем відчуваєття білю. Наприклад, при ушкодженні міжхребцевих дисків відбувається стискання спинномозкових нервів. При цьому болить та ділянка тіла, яка інервується защемленим нервом.
3) Відбитий (ірадіюючий, рефлекторний) біль – поширення больового відчуття із місця виникнення у віддалені ділянки.
Часто біль відбивається у ділянках периферії, які інервуються тим же сегментом спинного мозку, що і ушкоджений внутрішній орган. Якщо мова йде про поверхню шкіри, то біль відбивається у визначеному дерматомі (зону Захарівна-Геда для даного органа).
М о ж л и в і причини болей.
Біль викликають больові стимули - фактори, які обумовлюють сильне подразнення або ушкодження тканини. До них відносяться:
1) механічні больові стимули;
2) термічні больові стимули (температура вище 45°С);
3) хімічні больові стимули - ацетилхолін, серотонін, гістамін, кініни, простагландини Е2, субстанція Р, іони Н+ (при рН < 6), іони К+ (при концентрації понад 20 ммоль/л);
4) ушкодження нервових провідників.
Т е о р і ї механізмів болю.
1) Теорія інтенсивності. Її прихильники вважають, що в організмі відсутні спеціальні больові рецептори. Біль виникає в тому випадку, коли низькопорогові механо- і терморецептори стимулюються з інтенсивністю, яка перевищує визначений пороговий рівень. Якщо фактор діє із низькою або середньою інтенсивністю, то виникає тактильне або температурне відчуття, якщо ж інтенсивність висока - відчуття болю.
2) Теорія розподілу імпульсів. Її сутність полягає в тому, що больовий стимул викликає особливий хід нервових імпульсів, який відрізняється від поширення розрядів, характерних для дії неушкоджуючих факторів.
У цьому ряді стоїть "ворітна теорія" болю (Мелзак, Уолл), згідно якої велике значення у формуванні больових відчуттів належить желатинозній субстанції спинного мозку (substantia gelatinosa, SG).
Нейрони SG здійснюють пресинаптичне гальмування, блокуючи проходження імпульсів у нейрони задніх рогів спинного мозку по товстих і тонких нервових волокнах. За умов збудження нейронів SG відбувається пресинаптичне гальмування - "ворота" закриті. Якщо нейрони SG самі загальмовані, то пресинаптичне гальмування знімається - "ворота" відкриті.
Інтенсивна стимуляція товстих мієлінових нервових волокон викликає збудження нейронів SG - "ворота" закриваються, і проведення імпульсів у спинний мозок зменшується.
При інтенсивній стимуляції тонких немієлінових волокон, які проводять біль, відбувається гальмування нейронів SG і полекшення надходження імпульсів у задні роги спинного мозку.
3) Теорія специфічності. Передбачає існування специфічних больових рецепторів - ноціцепторів. Вони відповідають за сприйняття тільки інтенсивних стимулів і, таким чином, беруть участь у формуванні больових відчуттів.
Виділяють наступні види ноціцепторів:
- механочутливі ноціцептори (локалізуються у шкірі, скелетних м’язах);
- термочутливі ноціцептори - збуджуються при температурі вище 45°С (рецептори гарячого);
- полімодальні ноціцептори – збуджуються, як механічними, так і температурними больовими стимулами;
- хемочутливі ноціцептори - збуджуються хімічними болювими стимулами;
- вісцеральні ноціцептори - збуджуються розтяганням стінки гладком’язових органів або їх спастичним скороченням.
Хронічний біль - сильний, тривалий, виснажливий біль, який викликає страждання хворого.
В и д і л я ю т ь наступні форми хронічного болю:
1) Невралгія - больовий синдром, пов’язаний із порушеннями функції периферичних нервів при: а) вірусних інфекціях, б) авітамінозах, в) порушеннях кровообігу, г) розладах обміну речовин (цукровий діабет), д) хронічних отруєннях (солями важких металів, алкоголем). Особливо важкою є невралгія трійчастого нерва, яка проявляється приступами настільки сильного болю, що хворі не в змозі приймати їжу і розмовляти. Виникнення такого болю провокується дією дуже слабких подразників, наприклад, легким дотиком до шкіри обличчя.
2) Каузалгія - сильний пекучий біль, який виникає при ушкодженнях великих соматичних нервів. У хворого виникає відчуття, начебто на шкіру виливають окріп або прикладають руку до розпечених предметів. Навіть легкий дотик до шкіри, яка інервується ушкодженим нервом, викликає нестерпний біль. Біль може провокуватися несподіваними зоровими і слуховими подразниками.
3) Фантомний біль. Виникає після ампутації кінцівок - “болить” кінцівка, якої уже немає. При цьому біль дуже сильний і часто нестерпний.
4) Таламічний біль - важкий спонтанний біль у всій половині тіла з гіперпатією (суб’єктивним враженням підвищеної чутливості). Розвивається при ураженнях ядер таламуса.
Розрізняють наступні механізми формування хронічного болю:
1) Периферичні механізми:
а) хімічне подразнення і збільшення чутливості больових рецепторів (сенситизація ноціцепторів);
б) стискання нервів (наприклад, зсув хребцевих дисків → стискання корінців → больова імпульсація);
в) регенерація нервів (утворення потовщень - невром);
г) демієлінізація нервів (є провідним механізмом розвитку невралгії).
2) Периферично-центральні механізми:
а) патологічні рефлекси;
б) порушення балансу аферентних входів ("воротні" механізми болю);
в) зменшення гальмуючого впливу ретикулярної формації на "воротний" механізм болю;
г) денерваційна гіперчутливість.
3) Центральні механізми:
а) генерація патологічно посиленого збудження;
б) зняття гальмівного впливу кори головного мозку на таламічні ядра;
в) деаферентація нейронів;
г) зміни якості больових відчуттів.
Біль супроводжується загальними реакціями організму:
1) Емоційні реакції - депресія, апатія, загальне нездужання, хворобливий стан.
2) Вегетативні реакції - тахікардія, підвищення артеріального тиску, посилене потовиділення, гіперглікемія і гіперліпацидемія. Правда, тривалий біль, у тому числі і вісцеральний, супроводжується активацією парасимпатичної нервової системи, ознаками якої є брадикардія, падіння артеріального тиску.
3) Рухові реакції - рефлекторне скорочення скелетних м’язів (напруга м’язів черевної стінки при вісцеральних болях - симптом гострого живота, характерна поза при коліках).
4) Емоційно-больовий стрес. Біль і супровідні його механізми є причиною розвитку стресу.
5) Больовий шок. Біль є важливим патогенетичним механізмом розвитку різних видів шоку. Серед них травматичний, опіковий, кардіогенний, панкреатичний.
В організмі існують антиноціцептивні механізми.
Антиноціцептивними, або анальгезуючими називають природні механізми, які обмежують больові відчуття. Вони пригнічують проведення больових сигналів на всіх рівнях нервової системи, які беруть участь у формуванні відчуття болю.
В и д і л я ю т ь: а) нейрофізіологічні і б) нейрохімічні антиноцицептивні м е х а н і з м и.
• Нейрофізіологічні механізми пов’язані із групами нейронів, електрична стимуляція яких викликає пригнічення або повне вимикання діяльності різних рівнів аферентних систем, які передають ноціцептивну інформацію у вищі відділи мозку.
• Нейрохімічні механізми пов’язані із анальгезуючою дією хімічних речовин - нейромодуляторів. До них відносяться:
а) ендогенні опіоїдні пептиди (опіати) - енкефаліни, ендорфіни, дінорфіни, дерморфіни;
б) нейропептиди, які володіють вираженою дією на гладком’язові клітини кровоносних судин і внутрішніх органів - церулін, ксенопсин, фізалемін та ін.;
в) нейропептиди гіпоталамуса - вазопресин, окситоцин, соматостатин нейротензин.
Завдяки взаємодії нейрофізіологічних і нейрохімічниx механізмів в організмі функціонують 4 антиноціцептивних (анальгезуючих) системи:
1) Нейрона опіоїдна анальгуюча система. Її утворюють енкефалінергічні нейрони трьох рівнів: спинного, довгастого і середнього мозку.
2) Гормональна опіоїдна анальгезуюча система. Складається із 5 рівнів: а) спинний мозок, б) довгастий мозок, в) середній мозок, г) гіпоталамус, д) аденогіпофіз. В аденогіпофізі вивільняється β-ліпотропін, із якого утворюється β-ендорфін. Останній надходить у кров, досягає нервових структур і гальмує ноціцептивні нейрони спинного мозку і таламуса.
3) Нейрона неопіоїдна анальгезуюча система. Представлена моноаміноергічними структурами стовбура мозку: а) серотонінергічними, б) норадренергічними, в) дофамінергичними. Ці структури локалізуються в блакитній плямі, центральній сірій речовині середнього мозку.
4) Гормональна неопіоїдна анальгезуюча система. Активується при стрес-реакціях. Важливим її елементом є вазопресин, який виділяється клітинами гіпоталамуса в нейрогіпофіз, кров, спинномозкову рідину, а також безпосередньо в різні структури мозку: таламус, гіпокамп, мозочок, мигдалеподібне тіло, чорну субстанцію, ретикулярну формацію.
О с н о в н і принципи лікування болю:
1) Зменшення больової аферентації:
а) зменшення збудження рецепторів (імобілізація кінцівок, розслаблення м’язів);
6) збільшення порога больової чутливості (пригнічення утворення простагландинів, зменшення активності симпатоадреналової системи);
в) порушення проведення імпульсів від рецепторів по нервових провідниках (принцип анестезії).
2) Модуляція сенсорних входів. При збільшенні імпульсації по товстих нервових волокнах зменшується больова аферентація. Цей принцип лежить в основі фізичних методів знеболювання.
3) Активація ендогенних антиноціцептивних систем або імітація їх дії введенням фармакологічних антагоністів (введення наркотичних анальгетиків).
4) Пригнічення, руйнування або видалення центрів патологічної больової імпульсації в ЦНС.
5) Усунення психогенної больової патологічної домінанти.
На цих принципах засновані наступні методи знеболення:
1) Фармакологічні - застосування наркотичних і ненаркотичних анальгетиків, місцевоанестезуючих препаратів, антидепресантів і транквілізаторів.
2) Фізичні: а) черезшкірна електрична стимуляція нервів; б) глибоке прогрівання тканин; в) масаж; г) акупунктура (голковколювання).
3) Нейрохірургічні - видалення або руйнування структур ЦНС, які приймають участь у формуванні болю (наприклад, хордотомія - перерізання передньо-латерального тракту).
4) Психогенні: а) навіювання, б) гіпноз, в) аутогенні тренування.
Порушення рухової функції нервової системи. Експериментальне моделювання рухових розладів. Периферичні та центральні паралічі та парези: причини, механізми, прояви. Спінальний шок. Рухові порушення підкіркового походження. Порушення, пов'язані з ураженням мозочка. Судоми. Міастенія.
34.20. Як можна моделювати порушення рухових функцій нервової системи в експерименті?
В експериментальних дослідженнях використовують такі методи порушень рухових функцій нервової системи:
1) перетинання периферичних нервів і передніх корінців спинного мозку (відтворення периферичних паралічів);
2) ушкодження спинного мозку (відтворення спінального шоку);
3) перетинання стовбуру мозку між середнім і довгастим мозком (відтворення деце-ребраційної ригідності);
4) видалення мозочка;
5) перетинання пірамідних шляхів;
6) ушкодження структур екстрапірамідної системи;
7) видалення або електрична стимуляція рухових зон кори головного мозку.
34.21. Назвіть основні синдроми, що характеризують розлади рухової функції нервової системи.
1. Порушення нервово-м'язової передачі.
2. Периферичні паралічі й парези.
3. Центральні паралічі.
4. Синдром паркінсонізму.
5. Гіперкінетичні синдроми - гіперкінези.
6. Мозочковий синдром.
7. Судоми.
34.22. Назвіть причини і механізми розвитку порушень нервово-м'язової передачі. Що таке міастенія?
Основні причини порушення (блокади) нервово-м'язової передачі:
I. Механічне ушкодження нерва. Призводить до порушення проведення потенціалів дії до нервових терміналей і розладів аксоплазматичного транспорту.
II. Токсини і отрути. Серед них:
а) ботулінічний токсин;
б) а-бунгаротоксин (зміїна отрута);
в) кураре-екстракт, який отримують з рослин родів Strychnos і Chondodendron, що ростуть у Південній Америці. Здавна використовувався індіанцями як отрута для стріл;
г) інсектициди — засоби боротьби з комахами (хлорофос, дихлофос, карбофос); ґ) фосфорорганічні бойові отруйні речовини (хімічна зброя).
Ш. Фармакологічні агенти - міорелаксанти, інгібітори холінестерази.
IV. Спадкові фактори. Є причиною розвитку міастенії (Myasthenia gravis). Це захворювання, що виникає з частотою 1/20 000, виявляє себе м'язовою слабкістю і швидкою стомлюваністю у зв'язку з блокадою нервово-м'язової передачі. Вважають, що в основі міастенії - зменшення кількості ацетилхолінових рецепторів, можливо, обумовлене їх аутоімунним ушкодженням. Основні механізми порушень нервово-м'язової передачі:
1. Порушення проведення збудження до пресинаптичних нервових закінчень:
а) порушення анатомічної цілісності нерва;
б) порушення фізіологічної цілісності нерва - блокада проведення імпульсів у результаті зменшення амплітуди потенціалів дії або зменшення збудливості нервових волокон;
в) порушення енергетичного забезпечення нервових волокон;
г) деміелінізація нервів.
2. Порушення аксоплазматичного транспорту. Виникають при механічному ушкодженні нерва, порушеннях мікротрубочок, дефіциті енергії. У результаті розвиваються не тільки розлади нервово-м'язової передачі, але й порушення нервової трофіки (див. запит. 34.31).
3. Порушення синтезу і депонування ацетилхоліну в нервових терміналях. Можуть бути обумовлені:
а) дефіцитом вихідних продуктів синтезу ацетилхоліну - ацетил-КоА, холіну;
б) дефіцитом або зменшенням активності холін-ацетилтрансферази;
в) порушенням утворення синаптичних везикул;
г) порушенням транспорту ацетилхоліну з аксоплазми в синаптичні везикули..
4. Порушення вивільнення ацетилхоліну в сииаптичну щілину. Цей механізм лежить в основі дії ботулінічного токсину.
5. Порушення ацетилхолінових рецепторів постсинаптичної мембрани:
а) зменшення їхньої кількості;
б) оборотна (дія кураре) або необоротна (вплив а-бунгаротоксину) блокада ацетилхолінових рецепторів;
в) їх інактивація (десенситизація) і нечутливість постсинаптичних рецепторів до ацетилхоліну.
6. Розлади енергетичного обміну. При цьому страждають всі процеси, що потребують витрат енергії:
а) проведення потенціалів дії до пресинаптичних терміналей;
б) синтез і утворення в тілі нейрона компонентів нервових терміналей;
в) здійснення аксоплазматичного транспорту;
г) транспорт ацетилхоліну з аксоплазми в синаптичні везикули;
ґ) вивільнення вмісту синаптичних везикул у синаптичну щілину - екзоцитоз;
д) новоутворення синаптичних везикул з пресинаптичної мембрани - ендо-цитоз;
є) захоплення нервовими терміналями продуктів гідролізу ацетилхоліну (холіну, ацетату).
34.23. Що таке периферичні паралічі і парези? Чим вони характеризуються?
Параліч — це повна, а парез — часткова втрата довільних рухів. Периферичні паралічі виникають при ушкодженні периферичних рухових нервів і при дегенерації а-мотонейронів спинного мозку. Останнє характерно для розвитку поліомієліту.
Ознаки периферичних паралічів і парезів:
1) атонія (гіпотонія) м'язів— зменшення їхнього тонусу. Звідси ще одна назва — млявий параліч;
2) арефлексія (гіпорефлексія) — відсутність або ослаблення спинномозкових рефлексів, у здійсненні яких бере участь ушкоджений нерв або мотонейрон (розрив рефлекторної дуги);
3) атрофія м 'язів, обумовлена їхньою гіпофункцією.
34.24. Що таке центральні паралічі? Чим вони характеризуються?
Центральні паралічі виникають при ушкодженні центральних рухових низхідних шляхів. Найчастішими їх причинами є травми спинного мозку і розлади мозкового кровообігу (інсульти).
У розвитку основних ознак церебральних паралічів провідну роль відіграє зменшення гальмівних впливів з розташованих вище нервових центрів на а-мотонейрони спинного мозку. Цим пояснюють такі характеристики центрального паралічу:
1) гіпертонію — збільшення тонусу м'язів (спастичний параліч);
2) гіперрефлексію — посилення спинномозкових рефлексів (збільшення їхньої амплітуди і розширення зони, з якої їх можна викликати);
3) поява патологічних рефлексів (рефлекс Бабінського та ін.).
34.25. Що таке синдром паркінсонізму? Чим він обумовлений і чим виявляється?
Паркінсонізм є одним з найпоширеніших гіперкінетико-гіпертонічних синдромів.
У його патогенезі провідна роль, як вважають, належить недостатності дофа-мінергічних систем головного мозку. Відбувається руйнування патологічним процесом чорної субстанції, де утворюється дофамін.
У нормі дофамін із чорної субстанції по нігростріарних шляхах надходить у хвостате ядро. Там він є гальмівним медіатором, що пригнічує діяльність нейронів, які затримують і обмежують рухові акти (гальмується гальмування).
При руйнуванні чорної субстанції утворення дофаміну зменшується, у результаті чого збільшується активність нейронів хвостатого ядра і, отже, посилюється гальмування рухових актів — розвивається гіпокінезія.
Основні прояви паркінсонізму:
1) гіпокінезія- мала рухова активність, обличчя стає маскоподібним, жестикуляція бідною, рухи повільними. Ходьба характеризується дуже дрібними кроками і відсутністю природного хитання рук;
2) м 'язоваригідність (гіпертонус м'язів). Виявляється своєрідним опором пасивним рухам;
3) тремор - тремтіння пальців рук і кистей, а також нижньої щелепи.
34.26. Чим можуть виявлятися гіперкінетичні порушення рухових функцій центральної нервової системи?
Гіперкінетичні синдроми - гіперкінези - розвиваються в результаті уражень екс-трстірамідної системи. До них відносять:
1. Хорею — безладні мимовільні рухи з вираженим локомоторним ефектом, що супроводжуються гіпотонією м'язів (хорея - танок, або хвороба, святого Вітта). Цей синдром є прямою протилежністю паркінсонізму. У його основі морфологічні зміни в смугастому тілі (шкарлупі і хвостатому ядрі).
2. Атетоз - повільні тонічні скорочення м'язів, зовні схожі на "червоподібні" рухи, що, як правило, відбуваються в дистальних відділах рук.
3. Гемібалізм — швидкі розгонисті рухи рук, що нагадують кидання або штовхання м'яча.
34.27. Назвіть основні прояви рухових мозочкових порушень.
1. Атаксія — порушення координації рухів. Розрізняють статико-локомоторну і динамічну атаксію. У разі першої з них порушуються в основному стояння і ходьба, у разі другої - виконання кінцівками різних довільних рухів. Статшо-локомо-торна атаксія виникає при ураженнях черв'яка, а динамічна — при патологічних процесах у корі півкуль мозочка. До ознак атаксії відносять:
а) асинергію — порушення синергізму рухів: рухи виконуються не одночасно, а послідовно (розщеплення рухів);
б) дисметрію — рухи виконуються або в надлишковому, або в недостатньому обсязі.
2. Інтенційний тремор. Його нема в стані спокою, але він з 'являється під час руху руки, коли треба досягти якої-небудь мети, наприклад, доторкнутися до кінчика носа.
3. Гіпотонія м 'язів. У результаті розвивається слабкість мускулатури, швидка стомлюваність.
4. Ністагм — рухи очних яблук, що швидко повторюються.
5. Запаморочення.
6. Дефекти мови - скандована мова.
34.28. Що таке судоми? Які бувають види судом?
Судоми — це мимовільні скорочення скелетних м'язів, що мають характер нападів. Вони є особливим різновидом гіперкінезів.
Розрізняють клонічні і тонічні судоми. Клонічні характеризуються короткочасним скороченням і розслабленням окремих груп м'язів, що настають швидко одне
за одним. Для тонічних судом характерні тривалі скорочення м'язів, що створюють ефект "застигання" тулуба і кінцівок у яких-небудь вимушених позах.
Можливе поєднання різних компонентів судом, тоді мова йде про клонічно-то-нічні (переважає клонічний компонент) або тонічно-клонічні (більш виражені тонічні зміни) судоми.
За поширеністю судоми можуть бути місцевими і генералізованими.
Причиною судом, як правило, є надмірне збудження підкіркових структур головного мозку.
Залежно від механізмів розвитку нападів судом розрізняють:
1) судомну реакцію. Вона виникає епізодично на дію надзвичайного для даного організму подразника. Може розвиватися у будь-якої здорової людини, але частіше і легше виникає у людей з так званою судомною готовністю. У дітей віком до З років судомні реакції виникають у 4-5 разів частіше, ніж у дорослих. Причиною судомних реакцій можуть бути: гіпертермія, гіпокальціємія (тетанія), гіпоглікемія, гіпоксія, екзогенне отруєння та ін.;
2) судомний синдром. Виникає в умовах активного патологічного процесу в центральній нервовій системі. У його розвитку вирішальне значення має зменшення порогу судомної готовності і формування в рухових центрах головного мозку "патологічної домінанти" — осередку патологічного збудження;
3) епілепсія - хронічне захворювання, що характеризується повторними судомними і (або) психопатологічними нападами і нерідко зміною рис особистості. Етіологію і патогенез епілепсії ще не з'ясовано.
Порушення вегетативних функцій нервової системи, методи експериментального моделювання. Синдром вегетосудинної дистонії.
34.29. Які методи використовують для експериментального моделювання порушень вегетативної нервової системи?
I. Хірургічні методи: подразнення або руйнування центрів парасимпатичної і симпатичної нервової системи, перетинання вегетативних нервів, видалення вегетативних гангліїв.
II. Фармакологічні методи: введення в організм симпатолітиків, адреноблокаторів, холінолітиків (фармакологічна денервація).
III. Імунологічні методи: введення антитіл проти білків, що входять до складу гангліїв вегетативної нервової системи (наприклад, імунний симпатоліз).
34.30. Які конституціональні типи людей виділяють залежно від функціональної активності вегетативної нервової системи? Чим вони характеризуються?
Симпатикотонічна конституція. Переважає тонус симпатичної нервової системи. Характерна блідість і сухість шкіри, холодні кінцівки, блиск очей і м'який екзофтальм, нестійка температура тіла, схильність до тахікардії і підвищення артеріального тиску, закрепи, висока працездатність, особливо під вечір, ініціативність, тривожність, фізична витривалість, неспокійний сон, погана переносність сонця, тепла, шуму, яскравого світла, іноді неприємні відчуття у ділянці серця.
Ваготонічна конституція. Переважає тонус парасимпатичної нервової системи. Характерні холодна, волога, бліда шкіра, гіпергідроз (пітливість), гіперсалівація, яскравий червоний дермографізм, брадикардія, схильність до падіння артеріального тиску, дихальна аритмія, схильність до непритомностей, тенденція до збільшення маси тіла, апатичність, астенія, схильність до депресії, мала фізична витривалість, нерішучість, слабка ініціативність, висока чутливість, боязкість, більша працездатність у ранковий час.
34.31. Що таке синдром вегетосудинноїдистонії? Які його варіанти існують?
Синдром вегетосудинної дистонії (СВД) - це узагальнена назва порушень вегетативної нервової системи людини. Класифікація.